
Abstrak
Autofagi adalah proses katabolik yang dilakukan sel untuk mempertahankan homeostasis seluler melalui degradasi komponen sitoplasma yang tidak berfungsi, seperti protein salah lipat yang beracun dan organel yang rusak, di dalam lisosom. Proses ini merupakan proses multitahap yang diatur secara ketat oleh mekanisme penginderaan nutrisi, energi, dan stres. Autofagi memainkan peran penting dalam berbagai proses biologis, termasuk pengendalian kualitas protein dan organel, pertahanan terhadap infeksi patogen, metabolisme sel, dan pengawasan imun. Akibatnya, disfungsi autofagi dikaitkan dengan berbagai kondisi patologis. Peran autofagi dalam kanker bersifat kompleks dan dinamis. Bergantung pada konteksnya, autofagi dapat memiliki efek penekan tumor dan pro-tumorigenik. Sebaliknya, perannya lebih jelas didefinisikan dalam gangguan konformasi protein, di mana autofagi berfungsi sebagai mekanisme untuk mengurangi agregasi protein beracun, sehingga meningkatkan homeostasis seluler. Karena terapi berbasis autofagi memiliki potensi yang menjanjikan untuk pengobatan kanker dan gangguan konformasi protein, tinjauan ini akan menyoroti temuan dan kemajuan terbaru di bidang ini.
Singkatan
AATD , defisiensi alfa-1 antitripsin
AD , penyakit Alzheimer
ALS , sklerosis lateral amiotrofik
AMPK , kinase protein yang bergantung pada AMP
ATG , gen terkait autophagy
ATZ , Z alfa-1 antitripsin
CHOP , protein homolog C/EBP
CMA , autofagi yang dimediasi oleh pendamping
CQ , klorokuin
ER , retikulum endoplasma
ERAD , degradasi protein terkait ER
ERLAD , degradasi terkait ER ke lisosom
GABARAP , protein terkait reseptor asam gamma-aminobutyric
GOT1 , glutamat oksaloasetat transaminase 1
GOT2 , glutamat oksaloasetat transaminase 2
GWAS , studi asosiasi genom secara luas
HCC , karsinoma hepatoseluler
HCQ , hidroksiklorokuin
HD , penyakit Huntington
HDAC , histon deasetilase
HHC , hiperhomosisteinemia
HTT , Huntington
IL6 , interleukin-6
LAMP2A , membran terkait lisosom 2A
LIR , domain pengikat LC3
LSD , penyakit penyimpanan lisosomal
MAP1LC3 , protein terkait mikrotubulus
MAPT , protein terkait mikrotubulus Tau
ME1 , enzim malat 1
MiT/TFE , mikroftalmia/faktor transkripsi E
MTD , dosis maksimum yang dapat ditoleransi
mTOR , target mamalia rapamycin
mTORC , kompleks mTOR
Sel NK , sel imun pembunuh alami
PanIN , neoplasia intraepitel pankreas
PD , penyakit Parkinson
PDAC , adenokarsinoma duktal pankreas
PE , fosfatidiletanolamin
PI3K , fosfatidilinositol 3-kinase
PI3-P , fosfatidilinositol-3-fosfat
PPT1 , palmitoil-protein tioesterase 1
PrP , protein prion
SBMA , atrofi otot tulang belakang dan bulbar
SNAP 29 , protein terkait sinaptosomal 29
TCA , siklus asam trikarboksilat
TFEB , faktor transkripsi EB
TME , lingkungan mikro tumor
UPR , respons protein yang tidak terlipat
VAMP8 , protein membran terkait vesikel 8
VLDL , lipoprotein densitas sangat rendah
WIPI , pengulangan triptofan-asam aspartat (WD) domain interaksi fosfoinositida
Autofagi adalah proses katabolik dinamis dan terkonservasi secara evolusioner yang dengannya sel mengirimkan komponen sitoplasma ke lisosom untuk degradasi dan daur ulang. Bergantung pada mekanisme pengiriman muatan autofagi ke lisosom, tiga jenis autofagi telah dijelaskan: makroautofagi, mikroautofagi, dan autofagi yang dimediasi chaperone (CMA) [ [ 1 ] ]. Mikroautofagi melibatkan penyerapan langsung muatan sitoplasma melalui invaginasi membran lisosom, sedangkan CMA dicirikan oleh pengenalan protein substrat dengan motif seperti KFERQ oleh protein chaperone HSPA8, yang mengirimkannya ke lisosom untuk degradasi melalui membran terkait lisosom 2A (LAMP2A) [ [ 1 ] ]. Sebaliknya, selama makroautofagi (di sini disebut sebagai autofagi), kargo diasingkan oleh vesikel bermembran ganda yang disebut autofagosom, yang kemudian menyatu dengan lisosom untuk menghasilkan autolisosom, tempat kargo didegradasi. Proses autofagi dimediasi oleh serangkaian protein yang sangat terkonservasi di antara eukariota, yang dikenal sebagai protein terkait autofagi (ATG), yang penemuannya dilakukan melalui penyaringan genetik ragi yang dilakukan oleh lab Yoshinori Ohsumi [ [ 2 ] ]. Kontribusi perintis lainnya untuk pemahaman autofagi datang dari lab Daniel J. Klionsky. Saat mempelajari bagaimana jalur penargetan sitoplasma ke vakuola (Cvt) mengirimkan enzim aminopeptidase I langsung ke ekuivalen ragi dari lisosom, Klionsky dan timnya menemukan mekanisme perdagangan yang tidak biasa. Mekanisme ini berbeda dari jalur sekresi konvensional dan, pada kenyataannya, merupakan bentuk autofagi yang terspesialisasi. Investigasi lebih lanjut terhadap mutan jalur Cvt di Saccharomyces cerevisiae mengungkapkan adanya tumpang tindih dengan mutan autofagi dari laboratorium Yoshinori Ohsumi, sehingga meletakkan dasar untuk identifikasi dan karakterisasi gen terkait autofagi [ [ 3 ] ]. Para peneliti yang meneliti autofagi pada ragi menetapkan nomenklatur terpadu untuk gen terkait autofagi, yang menyediakan kerangka kerja standar untuk bidang tersebut [ [ 4 ] ]. Daftar lengkap gen terkait autofagi disertakan dalam Tabel S1 .
Autofagi diinduksi oleh berbagai rangsangan dan stres seluler. Awalnya dicirikan sebagai mekanisme adaptasi untuk mempertahankan kelangsungan hidup sel selama kekurangan nutrisi, autofagi juga terlibat dalam mempertahankan homeostasis seluler dengan menghilangkan organel yang rusak, protein yang salah lipat, dan organisme penyerang [ [ 5 , 6 ] ]. Inisiasi autofagi diatur oleh dua kinase yang dinamakan mammalian target of rapamycin (mTOR) dan AMP-dependent protein kinase (AMPK), yang merasakan ketersediaan nutrisi dan status energi seluler. Setelah diaktifkan, kedua kinase tersebut bertemu untuk memodulasi protein inisiasi autofagi melalui fosforilasi pada residu asam amino tertentu. Secara khusus, AMPK bertindak sebagai penginduksi autofagi, mengaktifkan protein kinase ATG ULK1 melalui fosforilasi Ser317, Ser77, dan Ser555, tergantung pada stres seluler yang menginduksi AMPK. Sebaliknya, mTOR menghambat autofagi dengan memfosforilasi ULK1 pada Ser757, sehingga memblokir aktivitasnya. Selain modifikasi pasca-translasi protein ATG, autofagi juga dapat diatur pada tingkat transkripsi. Contoh penting adalah faktor transkripsi EB (TFEB), pengatur utama biogenesis lisosomal, yang telah terbukti mengaktifkan program transkripsi yang mengendalikan langkah-langkah utama jalur autofagi sebagai respons terhadap kelaparan [ [ 7 ] ]. Aktivitas TFEB disetel dengan baik oleh kadar nutrisi ekstraseluler. Memang, setelah kekurangan nutrisi, TFEB didefosforilasi oleh kalsineurin, yang mengarah ke translokasi dan aktivasi nuklirnya [ [ 8 ] ]. Selain TFEB dan keluarga terkait dari faktor transkripsi/mikroftalmia E (MiT/TFE), banyak faktor transkripsi dan pengatur epigenetik lainnya telah terbukti meningkatkan ekspresi gen autofagi, seperti yang diulas di tempat lain [ [ 9 , 10 ] ]. Khususnya, mekanisme penting lain dari regulasi autofagi terjadi melalui respons protein yang tidak terlipat (UPR). Akumulasi protein yang salah lipat dalam retikulum endoplasma (ER) menyebabkan stres ER dan mengaktifkan jalur UPR, yang mengatur program transkripsi yang bertujuan untuk memulihkan homeostasis ER [ [ 11 ] ]. Secara khusus, dua faktor transkripsi jalur UPR, ATF4 dan protein homolog C/EBP (CHOP), telah terbukti mengatur transkripsi banyak gen ATG untuk meningkatkan pembersihan kesalahan pelipatan protein toksik dalam ER melalui autofagi [ [ 12 ] ].
Autofagi dan modulasinya telah terlibat dalam berbagai patologi manusia. Mengingat potensi terapi berbasis autofagi yang menjanjikan dalam mengobati kanker dan gangguan konformasi protein, tinjauan ini akan berfokus pada topik-topik ini, dengan menekankan perkembangan dan penemuan terkini. Secara khusus, tinjauan ini bertujuan untuk merangkum pengetahuan terkini tentang peran autofagi dalam kanker yang diinduksi RAS dan dalam gangguan konformasi protein yang memengaruhi hati dan otak. Uji klinis yang sedang berlangsung yang mengeksplorasi modulasi autofagi sebagai strategi terapeutik untuk pengobatan penyakit ini juga akan dibahas.
Mekanisme molekuler autofagi
Secara mekanistis, proses autofagi dapat dibagi menjadi beberapa langkah, termasuk pembentukan fagofor, pemanjangan membran isolasi, penutupan autophagosom, dan fusi dengan lisosom. Inisiasi autofagi dimediasi oleh kompleks ULK1/2, yang meliputi ULK1, ATG13, ATG101, dan FIP200/RB1CC1 (selanjutnya disebut FIP200). Kompleks ini terlibat dalam pembentukan fagofor dengan memfosforilasi dan merekrut kompleks fosfatidilinositol 3-kinase (PI3K) kelas III yang terdiri dari BECLIN 1, VPS34, ATG14, dan p115/USO1. Kompleks PI3K menghasilkan fosfatidilinositol-3-fosfat (PI3-P), yang dikenali oleh protein pengulangan domain fosfoinositida-berinteraksi (WIPI) triptofan-asam aspartat (WD). Secara khusus, WIPI2 telah terbukti mengikat ATG16L1, sehingga merekrut kompleks pemanjangan, yang mencakup ATG12, ATG5, dan ATG16L, ke lokasi perakitan fagofor [ [ 13 ] ]. Lokasi potensial nukleasi fagofor mencakup daerah retikulum endoplasma (ER) yang diperkaya dengan PI3-P, yang dikenal sebagai omegasom, serta lokasi kontak antara ER dan mitokondria, Golgi, dan membran plasma [ [ 14 – 18 ] ]. Setelah perakitan awal, fagofor diperpanjang oleh dua sistem konjugasi seperti ubiquitin. Secara khusus, ATG8 sitosolik diproses oleh protease sistein ATG4, yang membelah C-terminus ATG8, sehingga mengekspos residu glisin dan menghasilkan bentuk ATG8-I. Pada langkah berikutnya, ATG8-I dikonjugasikan dengan fosfatidiletanolamin (PE), yang mengarah pada pembentukan ATG8-II, yang dikaitkan dengan membran autophagosomal. Dalam proses ini, ATG7 dan ATG10 bertindak sebagai enzim mirip E1 dan E2, dan mengkonjugasikan ATG12 ke ATG5. Kompleks ATG12-ATG5 kemudian mengikat ATG16L1 untuk menghasilkan kompleks ATG12-ATG5-ATG16L1, yang memediasi konjugasi ATG8 dengan PE bekerja sama dengan ATG3. Pada mamalia, ada tujuh protein ATG8 berbeda yang termasuk dalam dua keluarga: protein terkait mikrotubulus (MAP1LC3) dan subfamili protein terkait reseptor asam gamma-aminobutyric (GABARAP). Protein-protein ini berfungsi sebagai perancah untuk merekrut protein lain, seperti adaptor untuk autophagy selektif. Memang, proses autofagi dapat mengenali muatan spesifik secara selektif melalui interaksi antara protein ATG8 pada membran autophagosomal dan protein reseptor seperti p62/SQSTM1 (selanjutnya disebut p62), yang terdapat pada muatan. Protein reseptor ini dicirikan oleh keberadaan domain pengikat LC3 (LIR), yang memungkinkan mereka berinteraksi dengan LC3 (ATG8). Beberapa jenis autofagi selektif telah dijelaskan, termasuk mitophagy (mitokondria), ER-phagy (retikulum endoplasma), aggrephagy (agregat protein), pexophagy (peroksisom), dan banyak lagi [ [ 19 , 20 ]]. Sumber lipid untuk pemanjangan dan pematangan autofagosom belum sepenuhnya dipahami. Namun, telah ditunjukkan bahwa membran sel yang berasal dari membran plasma, endosom, dan Golgi memasok lapisan ganda lipid ke fagofor yang baru lahir, yang memungkinkan perluasannya dalam proses yang melibatkan ATG9, satu-satunya protein ATG transmembran [ [ 21 , 22 ] ]. Langkah terakhir melibatkan penutupan fagofor dan fusi dengan lisosom. ATG2 telah ditunjukkan untuk mengatur penutupan autofagosom [ [ 23 ] ], sementara fusi autofagosom dengan lisosom dimediasi oleh beberapa protein, termasuk sintaksin 17 (STX17) dan protein terkait sinaptosom 29 (SNAP29) pada autofagosom dan protein membran terkait vesikel 8 (VAMP8) pada lisosom [ [ 24 ] ]. Isi autofagolisosom akhirnya didegradasi oleh hidrolase asam lisosomal dan dilepaskan ke sitoplasma untuk didaur ulang.
Autofagi dalam kanker
Peran autofagi dalam kanker bersifat kompleks dan dinamis, tergantung pada konteksnya, yang mencakup faktor-faktor seperti perubahan genetik dan stadium tumor. Penting juga untuk membedakan antara peran intrinsik autofagi dalam sel tumor dan fungsi autofagi non-sel-otonom dalam lingkungan mikro tumor (TME). Data terkini mendukung peran autofagi sebagai penekan tumor pada tahap awal tumorigenesis, sementara pada tumor yang sudah terbentuk, autofagi telah terbukti meningkatkan kelangsungan hidup sel kanker. Misalnya, autofagi membantu sel kanker untuk mengatasi kebutuhan metabolik dan stres dalam TME (misalnya, hipoksia), dan untuk memperoleh resistensi kemoterapi.
Fungsi autofagi untuk menekan tumor
Bukti pertama yang menyoroti peran autophagy sebagai mekanisme untuk memblokir tumorigenesis datang dari karya lab Beth Levine, yang menunjukkan bahwa BECLIN 1 bertindak sebagai penekan tumor pada model tikus [ [ 25 , 26 ] ]. Ablasi genetik penuh Becn1 telah ditemukan mematikan secara embrionik pada tikus, sementara tikus dengan penghapusan monoalel ( Becn1 +/− ) menunjukkan insiden limfoma dan karsinoma spontan yang lebih tinggi di paru-paru, hati, dan jaringan mamae, mengidentifikasi BECLIN 1 sebagai penekan tumor haplo-insufficient [ [ 27 ] ]. Efek tumorigenik yang diamati pada tikus Becn1 +/− tidak hanya dikaitkan dengan autophagy yang terganggu, tetapi juga dengan cacat pada trafficking endosom-lisosom. Penelitian sebelumnya telah menunjukkan bahwa defisiensi BECLIN 1 mengganggu pengangkutan reseptor substrat tirosin kinase (HGS) yang dimediasi oleh faktor pertumbuhan hepatosit dan merusak lokalisasi membran E-kadherin (CDH1) [ [ 28 , 29 ] ]. Penelitian genetik tikus lebih lanjut termasuk knockout Atg7 dan Atg5 juga mendukung peningkatan inisiasi tumor ketika autofagi terganggu [ [ 30 ] ]. Memang, tikus Atg7 −/− spesifik hati mengembangkan adenoma hati jinak, yang menunjukkan akumulasi p62, stres oksidatif, dan respons kerusakan DNA. Selain itu, ukuran tumor berkurang setelah penghapusan p62 secara bersamaan , yang menunjukkan bahwa akumulasi p62 berkontribusi pada perkembangan tumor. Penelitian lain juga mendukung potensi hubungan mekanistik antara perkembangan tumor dan reseptor autofagi p62. Dalam model adenokarsinoma paru yang diinduksi RAS, p62 diperlukan untuk perkembangan tumor dengan mempertahankan sinyal NF-kB [ [ 31 ] ]. Lebih jauh lagi, penekanan p62 dalam model autofagi yang rusak mencegah stres oksidatif, ketidakstabilan genomik, dan perkembangan tumor [ [ 32 ] ]. Yang penting, akumulasi p62 setelah hilangnya autofagi telah terbukti menginduksi karsinoma hepatoseluler dengan mengisolasi KEAP1, yang mengarah pada aktivasi faktor transkripsi NRF2 yang terus-menerus, yang pada gilirannya mendorong perkembangan tumor [ [ 33 ] ]. Penelitian lain lebih lanjut mendukung kontribusi sumbu p62-KEAP1-NRF2 dalam pertumbuhan kanker [ [ 34 – 37 ] ].
Selain peran perubahan genetik pada perkembangan tumor dalam pengaturan praklinis, studi lain menyoroti peran autofagi yang bergantung pada konteks dalam kanker. Dalam model tikus adenokarsinoma pankreas (PDAC) yang didorong oleh mutasi Kras , telah ditemukan bahwa peran autofagi bergantung pada status penekan tumor TP53 (selanjutnya disebut p53). Penghapusan Atg7 pada tikus yang membawa Kras mutan mengurangi perkembangan neoplasia intraepitel pankreas prakanker (PanIN) menjadi PDAC. Namun, hilangnya p53 secara bersamaan pada tikus mutan Kras yang dihapus untuk Atg7 menyebabkan perkembangan tumor pankreas yang lebih agresif, disertai dengan peningkatan kadar perantara jalur glikolitik dan pentosa fosfat [ [ 38 ] ].
Meskipun ablasi gen autofagi ditemukan untuk mempromosikan inisiasi tumor dalam model praklinis, studi pasien manusia telah menunjukkan bahwa gen mesin autofagi jarang bermutasi pada kanker manusia [ [ 39 ] ]. Selain itu, hilangnya BECN1 pada tumor manusia tampaknya merupakan perubahan penumpang, mengingat kedekatannya dengan BRCA1 pada kromosom 17q21 [ [ 40 ] ]. Namun, sebuah studi korelasi menunjukkan bahwa ekspresi BECN1 dikaitkan dengan prognosis yang buruk pada kanker payudara estrogen-negatif [ [ 41 ] ]. Selain itu, amplifikasi kromosom 5q yang terkait dengan perkembangan karsinoma sel ginjal sel jernih telah terbukti menyebabkan pertumbuhan tumor melalui ekspresi berlebihan onkogen p62 [ [ 42 ] ].
Secara mekanistis, autofagi menyediakan fungsi anti-tumor melalui pembuangan protein salah lipat yang beracun, pembersihan organel yang tidak berfungsi (misalnya, mitokondria yang rusak oleh spesies oksigen reaktif), dan pemeliharaan stabilitas genom. Memang, model tikus dengan ablasi genetik gen autofagi menunjukkan peningkatan disfungsi mitokondria, stres metabolik dan oksidatif, kerusakan DNA, dan ketidakstabilan genom [ [ 32 , 43 – 45 ] ]. Yang penting, setelah kehilangan autofagi, akumulasi p62 memungkinkan sel kanker untuk mengatasi stres oksidatif melalui pensinyalan NRF2. Selain perannya dalam pemeliharaan homeostasis redoks, NRF2 juga terbukti menyebabkan pemrograman ulang metabolik dan meningkatkan proliferasi sel dengan mengarahkan glukosa dan glutamin ke jalur anabolik [ [ 46 ] ]. Peran penting lain dari autofagi dalam kanker adalah interaksi dengan jalur apoptosis. Misalnya, protein BCL-2 adalah pengatur utama yang menentukan autofagi dan apoptosis. Protein-protein ini berfungsi dengan mengikat dan menahan molekul-molekul BAX/BAK proapoptotik, sehingga menjaga integritas membran luar mitokondria. Mereka juga berinteraksi dengan BECLIN 1 melalui domain BH3-nya. Ekspresi berlebihan dari faktor-faktor tersebut atau modifikasi pasca-translasi dapat mengganggu kompleks protein BECLIN 1/BCL-2, yang menginduksi autofagi atau kaskade apoptosis. Selain itu, kaspase memediasi pembelahan BECLIN 1, melepaskan fragmen C-terminal BECLIN 1 yang terlokalisasi ke mitokondria dan menginduksi aktivasi faktor-faktor proapoptotik [ [ 47 , 48 ] ]. Autofagi juga secara mekanistik terkait dengan penekan tumor p53. p53 adalah sensor intraseluler dari stres genotoksis yang mengendalikan proliferasi sel dengan menginduksi penangkapan siklus sel dan apoptosis sebagai respons terhadap stres yang berbeda termasuk kerusakan DNA, hipoksia, dan aktivasi onkogen [ [ 49 ] ]. Peran p53 pada autofagi bergantung pada lokalisasi subselulernya [ [ 50 ] ]. Ketika berada di sitoplasma, p53 memberikan fungsi penghambatan pada autofagi melalui interaksi protein-protein dengan FIP200, yang menghasilkan penghambatan kompleks inisiasi ULK1/2 [ [ 51 , 52 ] ]. Namun, setelah terjadi kerusakan seluler, p53 bertranslokasi ke nukleus, tempat ia mengatur transkripsi berbagai gen yang terlibat dalam autofagi. Salah satunya adalah modulator autofagi yang diatur oleh kerusakan ( DRAM1 ) [ [ 53 ] ], yang terlibat dalam pematangan LC3, pengasaman lisosom, dan degradasi [ [ 54 ] ], dan juga terlibat dalam apoptosis yang dimediasi p53 [[ 55 ] ].
Peran autofagi dalam mendorong tumor
Pada tahap lanjut perkembangan tumor, autofagi memainkan peran pro-kelangsungan hidup dengan memberikan plastisitas metabolik pada sel tumor yang mengalami stres karena tingkat proliferasi yang tinggi dan paparan hipoksia, yang diakibatkan oleh vaskularisasi yang buruk [ [ 56 ] ] (Gbr. 1A ). Melalui daur ulang nutrisi intraseluler, autofagi mendukung permintaan metabolik tumor yang berproliferasi dengan menyediakan metabolit yang diperlukan untuk biosintesis dan produksi energi. Misalnya, degradasi karbohidrat menjadi gula dapat memicu glikolisis; asam amino yang berasal dari degradasi protein autofagi dapat memasuki siklus asam trikarboksilat (TCA) di banyak titik, yang menopang metabolisme mitokondria; dan lipid dapat diubah menjadi asam lemak, yang digunakan untuk menghasilkan Asetil-KoA, sehingga memberi makan siklus TCA [ [ 57 , 58 ] ] (Gbr. 1A ). Selain itu, kemampuan sel tumor untuk mengamankan ketersediaan nutrisi yang cukup dengan mengaktifkan jalur daur ulang seperti autofagi terkait dengan perubahan genetik spesifik yang menginduksi pemrograman ulang metabolik. Misalnya, pada PDAC, mutan onkogenik KRAS mengaktifkan program transkripsi MiT/TFE untuk menginduksi autophagy, yang memasok sel kanker dengan asam amino spesifik yang dibutuhkan untuk pertumbuhan, seperti glutamin [ [ 57 , 59 , 60 ] ]. Khususnya, tumor PDAC, tempat mutan onkogenik KRAS umum, bergantung pada glutamin untuk bertahan hidup, karena menyediakan sumber karbon dan nitrogen untuk bahan bakar siklus TCA dan biosintesis nukleotida, masing-masing [ [ 61 , 62 ] ]. Selain itu, pada tumor PDAC, mutan KRAS meningkatkan glutaminolisis untuk memungkinkan pembentukan NADPH dan menjaga keseimbangan redoks melalui jalur non-kanonik yang melibatkan glutamat oksaloasetat transaminase 1 dan 2 (GOT1 dan GOT2) dan enzim malat 1 (ME1) [ [ 63 ] ]. Mengingat temuan-temuan yang menghubungkan pensinyalan RAS onkogenik, pemrograman ulang metabolik, dan autofagi, penghambatan kombinasi jalur RAS dan autofagi telah dieksplorasi baik dalam pengaturan praklinis maupun uji klinis, seperti yang dibahas di bagian berikutnya.
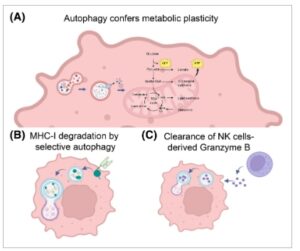
Gbr. 1
Buka di penampil gambar
Kekuatan Gambar
Contoh mekanisme yang digunakan autofagi untuk meningkatkan perkembangan tumor. (A) Autofagi adalah jalur katabolik yang mendaur ulang nutrisi, sehingga menghasilkan plastisitas metabolik. (B) Melalui degradasi molekul MHC-I, autofagi mendukung penghindaran kekebalan kanker dengan mencegah pengenalan sel T. (C) Autofagi membantu sel kanker untuk mendegradasi Granzyme B yang disekresikan oleh sel Natural Killer (NK). Dibuat di BioRender.
Peran pro-tumor penting lainnya dari autofagi adalah kemampuannya untuk memberikan resistensi terhadap berbagai regimen terapi anti-neoplastik [ [ 64 , 65 ] ]. Misalnya, penghambatan autofagi menggunakan klorokuin atau dengan merobohkan Atg5 menggunakan shRNA (short hairpin RNA) meningkatkan kemampuan kemoterapi obat alkilasi untuk menginduksi kematian sel tumor dalam model limfoma yang diinduksi MYC [ [ 66 ] ]. Penghambatan autofagi juga menunjukkan efek anti-proliferatif sinergis dalam kombinasi dengan terapi yang ditargetkan (misalnya, trametinib, everolimus, dan sorafenib) dalam berbagai model kanker, termasuk kanker pankreas yang diinduksi RAS, melanoma, dan kanker kolorektal bermutasi BRAF [ [ 67 – 69 ] ]. Di luar contoh-contoh yang disebutkan, penelitian lain lebih lanjut mendukung fungsi sitoprotektif autofagi sebagai respons adaptif juga terhadap terapi radiasi [ [ 70 – 75 ] ].
Bukti kuat mendukung peran autofagi dalam proses penghindaran imun sel kanker, di mana autofagi membantu sel tumor untuk lolos dari pengenalan sel imun. Salah satu mekanisme eliminasi sel tumor oleh imunitas adaptif didasarkan pada interaksi antara molekul MHC-I, yang mengandung neoantigen tumor, dan reseptor sel T pada permukaan limfosit T sitotoksik. Pada sel adenokarsinoma duktal pankreas, autofagi selektif ditemukan untuk mendorong degradasi molekul MHC-I melalui NBR1, sehingga mencegah pengenalan oleh sel imun [ [ 76 ] ] (Gbr. 1B ). Studi lain menunjukkan bahwa aktivasi autofagi di bawah hipoksia menyebabkan degradasi granzim B yang dilepaskan oleh sel Natural Killer (NK), membuat sel tumor hipoksia kurang sensitif terhadap pembunuhan yang dimediasi NK [ [ 77 ] ] (Gbr. 1C ).
Bahasa Indonesia: Selain fungsi intrinsiknya pada sel tumor, autofagi juga berperan dalam lingkungan mikro tumor (TME). Misalnya, autofagi mengatur produksi dan sekresi kemokin dan sitokin, yang pada gilirannya meningkatkan migrasi dan invasi sel kanker atau memodulasi infiltrasi sel T dan pengawasan imun. Dalam satu contoh, Valencia dan rekan penulis menunjukkan bahwa reseptor autofagi p62 bertindak sebagai penekan tumor pada fibroblas stroma. Secara khusus, penghapusan p62 pada fibroblas stroma mengurangi ekspresi mTORC1/c-MYC dan jalur detoksifikasi metabolik, yang mengarah pada peningkatan stres oksidatif dan produksi interleukin-6 (IL6), yang meningkatkan proliferasi dan invasi sel kanker prostat [ [ 78 ] ]. Autofagi juga ditemukan memfasilitasi sekresi sitokin pro-invasif IL6 di TME dalam model kanker yang digerakkan oleh RAS [ [ 79 ] ]. Selain itu, penghapusan Fip200 pada sel epitel payudara telah terbukti mengurangi inisiasi dan perkembangan kanker payudara dengan meningkatkan sekresi kemokin, yang pada gilirannya menyebabkan peningkatan infiltrasi sel T efektor CD8 + yang berkontribusi pada pengawasan imun kanker [ [ 80 ] ].
Menargetkan autophagy untuk pengobatan kanker
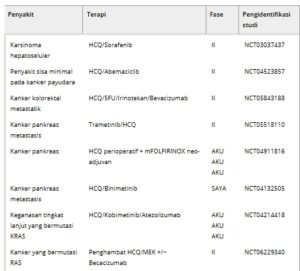
Jenis tumor tertentu, seperti tumor dengan jalur RAS dan RAF mutan, sangat bergantung pada autofagi bahkan dalam kondisi basal dan mungkin lebih sensitif terhadap penghambatannya [ [ 81 – 83 ] ]. Misalnya, tumor PDAC, di mana mutasi KRAS sangat sering terjadi, menunjukkan peningkatan autofagi basal dan sensitif terhadap penghambatan autofagi. Memang, knockdown genetik ATG5 dan penghambatan farmakologis autofagi (misalnya, Klorokuin) menurunkan pertumbuhan tumor secara in vitro dan in vivo [ [ 81 , 83 ] ]. Selain itu, sel tumor otak dengan mutasi BRAF V600E menunjukkan tingkat autofagi yang tinggi, dan penghambatan autofagi bersinergi dengan penghambat RAF vemurafenib untuk membunuh sel kanker [ [ 84 ] ]. Temuan ini menunjukkan bahwa penghambatan autofagi dapat menjadi strategi potensial untuk mengatasi resistensi kemoterapi. Autofagi dapat dihambat pada beberapa tahap, meskipun satu-satunya penghambat autofagi yang sejauh ini disetujui oleh Badan Pengawas Obat dan Makanan (FDA) untuk penggunaan klinis adalah klorokuin (CQ) dan hidroksiklorokuin (HCQ), yang menghambat tahap akhir autofagi. CQ ditemukan pada tahun 1934 oleh Hans Andersag dan rekan-rekannya di laboratorium Bayer, sementara turunannya, HCQ, dikembangkan pada tahun 1950-an dan berbeda dengan penambahan gugus hidroksil ekstra. Digunakan di klinik sebagai obat antimalaria, baik CQ maupun HCQ adalah basa lemah lipofilik yang mudah melewati membran plasma dan lebih disukai terakumulasi dalam lisosom sel yang bersifat asam. Pada pH lisosom, mereka sebagian besar hadir dalam bentuk terprotonasi, yang kurang permeabel terhadap membran, sehingga menjebak obat dalam lisosom. Hal ini menyebabkan peningkatan pH lisosom, menghambat protease lisosom dan fungsi lisosom secara keseluruhan [ [ 85 ] ]. Penelitian lebih lanjut mengidentifikasi palmitoyl-protein thioesterase 1 (PPT1) sebagai target molekuler yang dibagi di seluruh CQ dan turunannya menggunakan strategi penarikan fotoafinitas in situ [ [ 86 , 87 ] ]. Saat ini, banyak uji klinis sedang menyelidiki kemanjuran penghambat autofagi dalam kombinasi dengan rejimen anti-tumor lainnya, termasuk kemoterapi, terapi target, dan terapi radiasi [ [ 88 , 89 ] ]. Sebuah penyelidikan terhadap istilah “autofagi” dan “kanker” di situs web ClinicalTrials.gov menghasilkan 129 penelitian, yang 35 di antaranya masih merekrut peserta atau aktif. Beberapa dari uji klinis ini tercantum dalam Tabel 1Bukti klinis awal yang mendukung penggunaan penghambatan autofagi untuk meningkatkan hasil terapi datang dari uji coba kecil yang melibatkan pasien dengan glioblastoma. Kelompok pasien yang diobati dengan CQ dalam kombinasi dengan radiasi dan agen alkilasi Temozolomide memiliki kelangsungan hidup rata-rata yang diperpanjang secara signifikan dibandingkan dengan kontrol (33 bulan vs 11 bulan) [ [ 90 ] ]. Studi lebih lanjut mengevaluasi keamanan dan kemanjuran radiosensitisasi menggunakan CQ dalam kombinasi dengan radiasi seluruh otak untuk pengobatan metastasis otak. Meskipun CQ tidak meningkatkan kelangsungan hidup secara keseluruhan, studi-studi ini menunjukkan bahwa CQ ditoleransi dengan baik dan meningkatkan kontrol tumor intrakranial, yang menjamin penyelidikan lebih lanjut [ [ 91 , 92 ] ]. Uji klinis berikutnya menggunakan HCQ dalam kombinasi dengan rejimen terapi lain untuk mengkorelasikan parameter farmakokinetik-farmakodinamik dengan penghambatan autofagi. Uji klinis fase awal ini dilakukan pada pasien dengan berbagai jenis tumor, termasuk kanker paru-paru, melanoma, glioblastoma, dan mieloma refrakter. Dosis maksimum yang ditoleransi (MTD) HCQ bervariasi tergantung pada terapi bersamaan yang digunakan. Toksisitas pembatas dosis umum dalam uji coba ini termasuk toksisitas gastrointestinal dan kelelahan, dan, dalam beberapa kasus, juga mielosupresi. Khususnya, neurotoksisitas yang diinduksi HCQ tidak ada, yang kontras dengan prediksi dari studi model tikus [ [ 93 ] ]. Contoh uji coba ini termasuk studi fase I penghambat histone deacetylase (HDAC) Vorinostat dengan HCQ pada tumor padat, yang menetapkan bahwa HCQ 600 mg setiap hari dikombinasikan dengan Vorinostat 400 mg setiap hari adalah MTD dan rejimen fase II yang direkomendasikan [ [ 94 ] ]. Contoh lain adalah studi fase I Erlotinib dan HCQ pada kanker paru non-sel kecil lanjut yang menunjukkan bahwa HCQ dengan atau tanpa Erlotinib aman dan ditoleransi dengan baik, yang mendefinisikan dosis fase II HCQ yang direkomendasikan 1000 mg bila diberikan dalam kombinasi dengan Erlotinib 150 mg [ [ 95 ] ]. Uji coba yang menggabungkan HCQ 600 mg dua kali sehari dengan inhibitor mTOR Temsirolimus 25 mg setiap hari pada pasien dengan tumor padat juga menemukan bahwa kombinasi ini aman dan dapat ditoleransi dengan baik [ [ 96 ] ]. Yang penting, studi fase I/II HCQ yang dikombinasikan dengan terapi radiasi dan Temozolomide (75 mg·m −2 setiap hari selama 6 minggu, diikuti oleh 150 mg·m −2 setiap hari selama lima hari berturut-turut setiap bulan selama 6 bulan) pada pasien dengan glioblastoma menemukan bahwa HCQ 600 mg setiap hari adalah MTD dalam kombinasi dengan rejimen ini, sementara pada dosis HCQ 800 mg setiap hari, pasien mengalami mielosupresi (3/3 pasien dengan trombositopenia tingkat 4 dan 2/3 pasien dengan neutropenia tingkat 4) [ [97 ] ]. Sebaliknya, uji klinis lain yang melibatkan HCQ tidak mengamati toksisitas yang membatasi dosis. Misalnya, uji fase I yang menggabungkan Temozolomide (150 mg·m −2 setiap hari selama 7 dari setiap 14 hari) dengan HCQ 1200 mg setiap hari tidak mengamati mielosupresi [ [ 98 ] ]. Uji kombinasi HCQ lainnya juga mencapai dosis HCQ 1200 mg setiap hari [ [ 96 , 99 ] ], yang menunjukkan bahwa pemberian dosis Temozolomide secara terus-menerus atau kombinasi dengan HCQ selama kemoradiasi menghasilkan lebih banyak toksisitas dalam kompartemen hematopoietik daripada Temozolomide intermiten. Dalam uji kanker pankreas, monoterapi HCQ tidak memiliki manfaat klinis [ [ 100 ] ]. Namun, HCQ neo-adjuvant dengan kemoterapi menghasilkan hasil positif pada pasien dengan adenokarsinoma pankreas yang dapat direseksi, termasuk peningkatan respons tumor dan penurunan kadar serum CA 19-9. Yang penting, pasien yang menunjukkan penghambatan autofagi yang efektif, dievaluasi dengan imunoblotting untuk penanda autofagi LC3-II dalam sel mononuklear darah perifer yang bersirkulasi, menunjukkan peningkatan kelangsungan hidup bebas penyakit dan kelangsungan hidup secara keseluruhan [ [ 101 , 102 ] ].
Tabel 1. Contoh uji klinis yang menargetkan autofagi untuk terapi kanker yang saat ini sedang merekrut pasien.
Meskipun hasil yang menjanjikan ini, beberapa pertanyaan terbuka masih ada. Misalnya, identifikasi biomarker untuk memprediksi pasien dan jenis tumor mana yang mendapat manfaat dari penghambatan autofagi masih kurang. Lebih jauh, CQ dan HCQ memerlukan konsentrasi tinggi untuk mengganggu autofagi secara efektif, dan tidak jelas apakah mereka dapat mencapai penghambatan yang cukup pada dosis yang digunakan dalam uji klinis. Oleh karena itu, ada upaya intensif untuk mengidentifikasi penghambat autofagi yang lebih spesifik dan kuat. Studi baru difokuskan pada penargetan tahap awal autofagi, termasuk pengatur inisiasi autofagi seperti VPS34 [ [ 103 – 105 ] ], ULK1/2 [ [ 106 – 108 ] ], dan ATG4B [ [ 109 ] ]. Selain itu, penghambat lisosomal generasi berikutnya sedang dalam pengembangan, seperti VATG-032 [ [ 110 ] ], Lys05 [ [ 111 ] ], dan DQ661 [ [ 86 ] ]. Agen lisosomotropik baru ini, yang dicirikan oleh dua cincin aminoquinoline yang dipisahkan oleh penghubung, telah menunjukkan spesifisitas dan potensi yang lebih besar dalam hal penghambatan tumor dan de-asidifikasi lisosomal dibandingkan dengan CQ dan HCQ dalam studi praklinis, bahkan bekerja sebagai agen tunggal. Sementara molekul-molekul kecil ini telah menunjukkan hasil yang menjanjikan dalam model praklinis, efek yang tidak sesuai target dapat membatasi kegunaannya, seperti dalam kasus inhibitor ULK1/2, yang juga menargetkan kinase lain [ [ 112 ] ]. Lebih jauh lagi, efek farmakologis pleiotropik dari inhibitor ini akan mempersulit upaya untuk menentukan peran spesifik autofagi dalam hasil terapeutiknya.
Autophagy dalam gangguan konformasi protein
Gangguan konformasi protein adalah kelas penyakit yang disebabkan oleh akumulasi protein salah lipat yang mengganggu fungsi seluler dan fisiologi jaringan, yang mengarah pada kondisi patologis [ [ 113 ] ]. Protein dapat gagal melipat ke konformasi tiga dimensi yang benar karena beberapa alasan, yang mengakibatkan akumulasi mereka dalam badan inklusi. Dalam bentuk agregat ini, protein resistan terhadap pembersihan dan dapat mengganggu fungsi sel normal dengan mengganggu transportasi RNA dan protein dan dengan mengisolasi protein normal, seperti chaperone, ke dalam endapan intraseluler yang tidak larut [ [ 114 , 115 ] ]. Dengan demikian, salah lipat protein tidak hanya menyebabkan hilangnya fungsi kanonik protein tetapi juga memperkenalkan sifat toksik baru melalui mekanisme perolehan fungsi. Dalam beberapa kasus, seperti penyakit prion, protein salah lipat dapat memperoleh kemampuan replikasi diri, penularan sel ke sel, dan infeksi lintas spesies. Dalam kebanyakan kasus, konfigurasi molekuler penyebab penyakit melibatkan peningkatan struktur sekunder β-sheet dan hubungan β-sheet [ [ 116 , 117 ] ]. Contoh gangguan konformasi protein meliputi penyakit Alzheimer (AD), penyakit Parkinson (PD), penyakit Huntington (HD), sklerosis lateral amiotrofik (ALS), dan lainnya. Faktor risiko untuk kondisi ini meliputi polimorfisme genetik, penuaan, dan faktor lingkungan seperti cedera otak traumatis dan infeksi virus. Faktor lain yang menyebabkan agregasi protein meliputi modifikasi pascatranslasi, seperti hiperfosforilasi protein MAPT (protein Tau terkait mikrotubulus), perubahan suhu atau pH, perubahan chaperone yang membantu pelipatan protein, peningkatan produksi protein, atau penurunan pembersihannya.
Mengingat perannya dalam pembuangan organel yang rusak (misalnya, mitokondria dan retikulum endoplasma) dan agregat protein toksik, autofagi memainkan peran sitoprotektif dalam menjaga homeostasis sel. Akibatnya, banyak penelitian telah mengeksplorasi potensi modulasi autofagi sebagai strategi terapeutik untuk gangguan konformasi protein.
Gangguan konformasi protein yang mempengaruhi otak
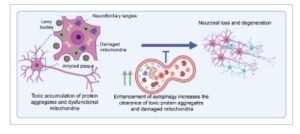
Ciri khas dari banyak penyakit neurodegeneratif adalah pembentukan progresif agregat protein yang tidak larut yang dikenal sebagai amiloid [ [ 118 ] ]. Istilah amiloid diperkenalkan oleh Rudolf Virchow pada tahun 1854 untuk menggambarkan kelainan jaringan makroskopis yang menunjukkan reaksi pewarnaan yodium positif, menyerupai pati [ [ 119 – 121 ] ]. Kemudian, ditemukan bahwa struktur amiloid tersusun dari protein, yang mengarah pada pengenalan gangguan konformasi protein. Fibril protein amiloid tersusun dari lembaran-β intermolekul yang berinteraksi erat dan berulang, membentuk struktur lembaran-β silang yang diwarnai positif dengan pewarna merah Kongo [ [ 122 , 123 ] ]. Mekanisme molekuler yang mendasari patogenesis banyak penyakit neurodegeneratif melibatkan pengendapan intraseluler fibril protein amiloid [ [ 116 , 121 ] ] (Gbr. 2 ). Misalnya, deposisi ekstraseluler protein amiloid-beta (Aβ) dan jalinan neurofibrilar intraseluler dari MAPT yang mengalami hiperfosforilasi merupakan ciri khas AD [ [ 124 ] ] (Gbr. 2 ). PD dicirikan oleh agregat alfa-sinuklein dalam badan Lewy, yang menyebabkan hilangnya neuron dopaminergik di substansia nigra dan menyebabkan disfungsi motorik progresif [ [ 125 ] ] (Gbr. 2 ). ALS juga dikaitkan dengan akumulasi protein yang salah lipat seperti SOD1, TDP-43, FUS, dan C9ORF72 [ [ 126 ] ]. Selain itu, beberapa kelainan neurodegeneratif bawaan langka melibatkan akumulasi intraseluler protein yang salah lipat dalam badan inklusi yang disebabkan oleh perluasan pengulangan poliglutamin (polyQ). Pengulangan glutamin ini dapat berkembang lintas generasi, dan ketika panjangnya melebihi ambang batas, terjadi hubungan lembaran untai β intermolekul, yang mengakibatkan agregat nuklir yang tidak larut dan manifestasi penyakit. Contoh penyakit tersebut meliputi HD (disebabkan oleh ekspansi polyQ pada huntingtin), atrofi otot spinal dan bulbar (SBMA) (disebabkan oleh ekspansi polyQ pada reseptor androgen), dan beberapa bentuk ataksia spinocerebellar (disebabkan oleh ekspansi polyQ pada protein ataxin) [ [ 127 ] ] . Khususnya, ensefalopati spongiform menular (TSE) adalah sekelompok penyakit neurodegeneratif langka yang ditandai dengan agregasi protein prion yang salah lipat (PrP) [ [ 128 ]]. Perubahan konformasi pada PrP melibatkan transisi dari struktur yang utamanya heliks menjadi struktur yang didominasi β-sheet, yang memberikan protein sifat replikasi diri dan penularan, yang memungkinkannya untuk berkembang biak menjadi prion normal. Konformasi prion patologis ini dapat menyebar antar individu atau bahkan antar spesies.
Gambar 2
Buka di penampil gambar
Kekuatan Gambar
Skema yang mengilustrasikan peningkatan autofagi sebagai strategi terapi potensial untuk pengobatan penyakit neurodegeneratif. Akumulasi agregat protein dan organel disfungsional intraseluler, seperti mitokondria, menyebabkan hilangnya neuron secara progresif dan neurodegenerasi. Stimulasi autofagi telah terbukti bersifat neuroprotektif, mencegah beban akumulasi protein toksik mencapai ambang batas patogenik. Dibuat dalam BioRender.
Autofagi memainkan peran penting dalam pembersihan fibril amiloid toksik dan organel yang rusak (Gbr. 2 ), sebagaimana didukung oleh banyak studi praklinis. Model tikus dengan penghapusan gen autofagi spesifik sistem saraf telah menunjukkan kelangsungan hidup yang berkurang dan neurodegenerasi progresif dini di seluruh area otak yang luas, dengan variabilitas tergantung pada gen yang ditargetkan, tahapan di mana autofagi terganggu, dan fungsi non-autofagi dari setiap target [ [ 129 – 132 ] ]. Yang penting, peningkatan autofagi melalui strategi seperti penghambatan mTOR atau aktivasi AMPK telah terbukti meningkatkan pembersihan protein salah lipat yang toksik dalam beberapa penelitian [ [ 133 – 138 ] ]. Misalnya, skrining berbasis mikroarray molekul kecil baru-baru ini mengidentifikasi empat senyawa penghubung yang berinteraksi dengan LC3 dan Huntingtin mutan (HTT), sehingga secara khusus memberikan HTT mutan ke autofagosom [ [ 139 ] ]. Studi lain menunjukkan perbaikan fenotipe penyakit pada model tikus HTT mutan dengan menggunakan molekul fusi yang terdiri dari QBP1 (yang secara spesifik mengikat traktus polyQ yang diperluas pada HTT mutan) dan motif pengikat HSPA8, yang mempromosikan autofagi yang dimediasi chaperone dari HTT mutan [ [ 140 ] ]. Selain bukti praklinis, mutasi genetik yang diwariskan pada gen terkait autofagi juga telah diidentifikasi pada pasien manusia. Misalnya, mutasi pada PRESENILIN 1 , yang menyebabkan AD familial, ditemukan mengganggu pengasaman autolisosom dan aktivasi katepsin, mengganggu pengangkutan subunit v-ATPase V0a1 ke lisosom [ [ 141 ] ]. Sebuah studi asosiasi genom-lebar (GWAS) yang mengimplikasikan protein adaptor clathrin, PICALM, dalam patogenesis AD menunjukkan bahwa hilangnya fungsi PICALM mengganggu endositosis protein VAMP, yang penting untuk pematangan autofagosom [ [ 142 , 143 ] ]. Mutasi pada faktor-faktor yang berhubungan dengan autofagi, seperti WIPI4/WDR45 , bertanggung jawab atas timbulnya Neurodegeneration with Brain Iron Accumulation (NBIA), kelainan gerakan neurologis bawaan langka yang ditandai dengan akumulasi zat besi yang tidak normal di otak dan degenerasi progresif sistem saraf [ [ 144 , 145 ] ]. Beberapa penyakit neurodegeneratif, seperti PD, dikaitkan dengan mitofagi, suatu bentuk autofagi selektif yang menargetkan mitokondria yang rusak. Mutasi pada PINK1 dan PRKN , dua protein yang terlibat dalam mitofagi, dikaitkan dengan PD awal resesif autosomal [[ 146 – 148 ] ]. ALS telah dikaitkan dengan mutasi pada gen reseptor autofagi, seperti p62/SQSTM1 [ [ 149 ] ], OPTN [ [ 150 ] ], dan UBIQUILIN 2 [ [ 151 ] ]. Selain itu, polimorfisme nukleotida tunggal dalam ATG7 telah dikaitkan dengan timbulnya penyakit lebih awal pada HD [ [ 152 ] ]. Akhirnya, penyakit penyimpanan lisosom (LSD), sekelompok kelainan bawaan langka yang menyebabkan kecacatan neurologis pada masa kanak-kanak atau dewasa [ [ 153 ] ], lebih lanjut menyoroti hubungan antara neurodegenerasi dan disfungsi autofagi-lisosom. Misalnya, pada penyakit Niemann-Pick tipe A, mutasi pada gen yang mengkode sfingomielinase asam memengaruhi komposisi dan fungsi lipid membran, mengganggu pergerakan mATG9 dan penutupan autofagosom [ [ 154 ] ]. Selain itu, faktor risiko genetik yang paling umum untuk PD adalah heterozigositas untuk mutasi patogenik pada enzim lisosomal glukoserebrosidase (GBA). Hilangnya aktivitas GBA menyebabkan akumulasi substratnya, glukosilseramida, di dalam lisosom, yang mengganggu autofagi karena disfungsi lisosomal [ [ 155 ] ].
Gangguan konformasi protein yang mempengaruhi hati
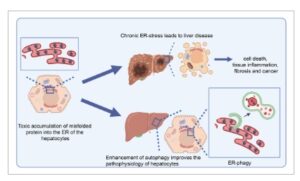
Gangguan konformasi protein yang memengaruhi hati terutama disebabkan oleh gangguan pada homeostasis retikulum endoplasma (ER) (Gbr. 3 ). Hati adalah organ yang sangat sintetis, tempat hepatosit (sel parenkim hati) melakukan berbagai fungsi metabolisme. Ini termasuk sintesis dan sekresi protein plasma, perakitan dan sekresi lipoprotein dan lipoprotein densitas sangat rendah (VLDL), biosintesis kolesterol, dan metabolisme xenobiotik dan lipid [ [ 156 ] ]. Sebagian besar proses biosintesis ini terjadi di dalam ER hepatosit, membuatnya sangat sensitif terhadap faktor genetik atau lingkungan (seperti infeksi virus) yang mengganggu homeostasis ER, yang berpotensi menyebabkan penyakit hati [ [ 157 ] ] (Gbr. 3 ). Berbagai rangsangan dapat menyebabkan disfungsi ER, termasuk stres oksidatif, perubahan komposisi lipid membran, hiperhomosisteinemia (HHC) yang menyebabkan protein N-homosisteinilasi, dan pembentukan agregat protein [ [ 156 ] ]. Yang terakhir melibatkan akumulasi protein yang salah lipat dalam ER hepatosit sebagai akibat dari kelainan bawaan monogenik, di mana mutasi pada gen yang mengkode protein kunci dapat menyebabkan pelipatan yang tidak tepat, seperti dalam kasus defisiensi antitripsin Alfa-1. Sebagai respons terhadap akumulasi protein yang salah lipat, ER mengaktifkan respons protein yang tidak terlipat (UPR), yang terdiri dari tiga transduser transmembran ER yang dikenal sebagai IRE1α, PERK, dan ATF6α. UPR diaktifkan untuk memulihkan homeostasis ER dan mengurangi beban protein yang salah lipat dengan memperluas kapasitas pelipatan ER, mengurangi sintesis protein, dan meningkatkan mekanisme pembuangan protein seperti degradasi protein terkait ER (ERAD) dan autofagi [ [ 158 ] ]. Autofagi yang diinduksi stres ER merupakan mekanisme kunci pengendalian kualitas ER, di mana fragmen ER yang rusak ditelan ke dalam autofagosom dan didegradasi setelah fusi dengan lisosom. Proses autofagi selektif ini, yang dikenal sebagai ER-fagi, berfungsi sebagai respons adaptif untuk mengurangi agregasi protein toksik dan memulihkan homeostasis ER [ [ 159 , 160 ] ] (Gbr. 3 ). Hingga saat ini, beberapa reseptor autofagi selektif yang terlibat dalam ER-fagi telah diidentifikasi, termasuk FAM134B [ [ 161 ] ], RTN3L [ [ 162 ] ], SEC62 [ [ 163 ] ], CCPG1 [ [ 164 ] ], ATL3 [ [ 165 ] ], dan TEX264 [ [ 166 ]]. Sebagian besar reseptor ini tidak memiliki domain lumen ER tetapi memiliki domain LIR (wilayah interaksi LC3), yang memungkinkan mereka berinteraksi langsung dengan LC3 atau GABARAP, yang pada gilirannya memfasilitasi perekrutan autofagosom ke ER. Kemungkinan reseptor ER-fagi ini juga berinteraksi dengan kofaktor lain yang membantu mengoordinasikan fungsinya, meskipun sifat pasti dari interaksi ini masih belum jelas. Selain itu, reseptor ER-fagi ini dapat mengalami modifikasi pascatranslasi yang mengatur aktivitasnya, yang selanjutnya memengaruhi kemanjuran ER-fagi. Investigasi lebih lanjut diperlukan untuk lebih memahami peran ER-fagi dan bagaimana ia diatur dalam konteks penyakit hati dan kondisi patologis lainnya. Pemahaman yang lebih rinci dapat mengarah pada strategi terapi baru yang menargetkan ER-fagi untuk mengurangi gangguan kesalahan pelipatan protein.
Gambar 3
Buka di penampil gambar
Kekuatan Gambar
Skema yang mengilustrasikan patogenesis penyakit hati akibat akumulasi protein toksik dan efek perlindungan autofagi. Akumulasi protein yang salah lipat dalam hepatosit menyebabkan kematian sel, peradangan hati, fibrosis, dan kanker. Peningkatan autofagi meningkatkan pembuangan protein toksik, sehingga memperbaiki ciri khas penyakit. Dibuat dalam BioRender.
Defisiensi antitripsin alfa-1 (AATD)
Prototipe kelainan konformasi protein yang mempengaruhi hati adalah defisiensi antitripsin Alfa-1 (AATD), yang disebabkan oleh mutasi pada gen SERPINA1 , yang mengkode untuk antitripsin alfa-1 (AAT) [ [ 113 , 167 ] ]. Berfungsi sebagai penghambat protease serin (Serpin), AAT terutama disintesis oleh hepatosit dan dilepaskan ke dalam sirkulasi untuk melindungi jaringan paru-paru dari degradasi proteolitik oleh enzim seperti elastase. Varian genetik yang paling umum pada pasien AATD adalah homozigositas untuk alel Z dari SERPINA1 , yang menghasilkan versi AAT yang bermutasi, yang dikenal sebagai antitripsin alfa-1 Z (ATZ). Protein mutan ini membawa mutasi salah arti (p.E342K), yang menyebabkannya salah lipat dan agregat, yang mengarah ke akumulasinya dalam ER hepatosit [ [ 168 ] ]. Seperti Serpin lainnya, AAT memiliki struktur molekul yang fleksibel, membuatnya rentan terhadap mutasi yang menyebabkan hubungan antar molekul dan pembentukan polimer, yang mengakibatkan hilangnya fungsi seluler secara bertahap [ [ 117 ] ]. Akumulasi ATZ dalam hepatosit mengakibatkan penyakit hati melalui mekanisme perolehan fungsi, yang mendorong peradangan hati, fibrosis, dan akhirnya karsinoma hepatoseluler (HCC) [ [ 169 ] ]. Studi menggunakan tikus transgenik PiZ, yang direkayasa untuk mengekspresikan alel Z, secara eksperimental telah menunjukkan mekanisme perolehan fungsi ini. Tikus-tikus ini menunjukkan kerusakan hati yang mencerminkan AATD manusia, dengan akumulasi ATZ hati, proliferasi hepatosit, peradangan, fibrosis, dan HCC, bahkan dengan adanya AAT tikus endogen [ [ 170 ] ]. Akumulasi ATZ intraseluler menyebabkan kelebihan beban ER, yang meningkatkan kerentanan terhadap stres ER, terutama bila diperparah dengan penghinaan seluler tambahan [ [ 171 ] ]. Degradasi ATZ terjadi terutama melalui sistem proteasom ubiquitin dan autofagi. Studi in vitro telah menunjukkan bahwa ATZ monomerik didegradasi oleh proteasom melalui jalur degradasi terkait ER (ERAD), sementara polimer ATZ sebagian didegradasi oleh autofagi, meskipun sebagian dari mereka bertahan sebagai inklusi yang tidak larut [ [ 172 ] ]. Sebuah studi baru-baru ini mengidentifikasi jalur degradasi terkait ER-ke-lisosom (ERLAD) alternatif, berbeda dari autofagi standar, yang mungkin bertanggung jawab untuk mendegradasi polimer ATZ yang resistan terhadap proteasom. Jalur ini menunjukkan bahwa degradasi ATZ mungkin tidak memerlukan penangkapan ER dalam autofagosom tetapi sebaliknya terjadi melalui transportasi vesikular yang dimediasi reseptor yang melibatkan kalneksin pendamping ER dan reseptor fagi ER FAM134B [ [ 173 ]]. Peran autofagi dalam AATD pertama kali disarankan ketika autophagosom yang melimpah diamati dalam sel mamalia yang mengekspresikan ATZ, hati tikus PiZ, dan hati pasien AATD [ [ 174 ] ]. Penelitian selanjutnya telah menunjukkan bahwa stimulasi autofagi, baik dengan molekul kecil (misalnya, karbamazepin) atau melalui transfer gen regulator autofagi TFEB, mengurangi akumulasi ATZ intraseluler dan mengurangi kerusakan hati pada tikus PiZ [ [ 175 , 176 ] ]. Berdasarkan temuan ini, uji klinis fase II (NCT01379469) menggunakan karbamazepin telah dieksplorasi untuk menyelidiki potensi modulasi autofagi sebagai strategi terapi untuk pengobatan penyakit hati pada pasien AATD (Tabel 2 ).
Tabel 2. Uji klinis yang telah selesai dan sedang berlangsung untuk gangguan konformasi protein yang memengaruhi hati dan otak.
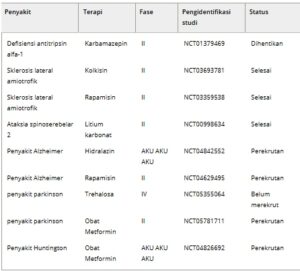
Menargetkan autophagy untuk pengobatan gangguan konformasi protein
Strategi terapi terkini untuk penyakit yang disebabkan oleh agregasi protein toksik berfokus pada pengurangan kadar protein agregat ini, baik dengan mengurangi ekspresi gen (misalnya, interferensi RNA) atau dengan merangsang mekanisme pembuangan protein seperti autofagi [ [ 177 ] ]. Peningkatan autofagi telah dieksplorasi melalui berbagai strategi, termasuk penggunaan modulator molekul kecil dan terapi gen.
Molekul kecil
Sejumlah modulator molekul kecil autofagi telah dikembangkan dan diuji dalam studi praklinis dan uji klinis. Senyawa-senyawa ini menargetkan berbagai langkah jalur autofagi, dari inisiasi (misalnya, kinase AMPK, mTOR, dan ULK1) hingga nukleasi (misalnya, VPS34), dan akhirnya, degradasi autofagosom dalam lisosom. Bagian ini akan membahas penginduksi molekul kecil paling umum yang menargetkan penghambatan mTOR dan aktivasi sinyal AMPK.
Aktivator AMPK
Trehalosa, disakarida glukosa yang terjadi secara alami, mengaktifkan autofagi secara independen dari kompleks mTORC1 [ [ 178 ] ]. Studi telah menunjukkan bahwa trehalosa menginduksi autofagi melalui aktivasi AMPK dengan menargetkan protein GLUT, keluarga transporter glukosa [ [ 179 ] ]. Lebih jauh lagi, trehalosa mempromosikan autofagi dengan mengaktifkan TFEB melalui penghambatan AKT1 [ [ 180 ] ]. Trehalosa telah diuji secara luas dalam model praklinis penyakit neurodegeneratif, termasuk model AD, PD, HD, penyakit Prion, dan ALS [ [ 137 , 138 , 181 – 186 ] ] dan saat ini sedang dievaluasi dalam uji klinis untuk pasien dengan PD (Tabel 2 ).
Metformin, obat antidiabetik yang disetujui FDA, juga merupakan penginduksi autofagi yang bergantung pada AMPK. Obat ini telah menunjukkan efek menguntungkan dalam model AD, HD, dan PD [ [ 187 – 190 ] ]. Resveratrol, stilbena alami yang ditemukan dalam anggur dan beri, memberikan fungsi neuroprotektif melalui aktivasi autofagi melalui pensinyalan AMPK/SIRT1 [ [ 191 , 192 ] ]. Aktivator AMPK tambahan seperti A769662 dan GSK621 juga telah dieksplorasi karena sifat neuroprotektifnya [ [ 193 – 196 ] ].
penghambat mTOR
Sebagai komponen inti dari dua kompleks protein yang berbeda, yaitu kompleks mTOR 1 dan 2 (mTORC1 dan 2), mTOR memainkan peran sentral dalam mengatur proses biologis fundamental seperti pertumbuhan, proliferasi, autophagy, imunitas, dan metabolisme [ [ 197 ] ]. Modulasi pensinyalan mTOR menggunakan inhibitor molekul kecil telah muncul sebagai jalan yang menjanjikan dalam penelitian medis, khususnya untuk terapi kanker dan gangguan neurologis. Inhibitor kinase mTOR dapat diklasifikasikan menjadi dua kategori: inhibitor non-ATP-kompetitif (generasi pertama) dan inhibitor ATP-kompetitif (generasi kedua) [ [ 198 ] ]. Inhibitor mTOR generasi pertama seperti rapamycin dan rapalog memberikan efeknya dengan mengikat protein FKBP12 dan membentuk kompleks yang menghambat mTORC1 tetapi tidak mTORC2 [ [ 199 ] ]. Rapamycin (juga dikenal sebagai Sirolimus) adalah makrolida yang diisolasi secara alami dari Streptomyces hygroscopicus , bakteri yang ditemukan di Pulau Paskah [ [ 200 ] ], yang menunjukkan kelarutan dan sifat farmakokinetik yang buruk. Untuk mengatasi keterbatasan ini, penelitian tambahan mengarah pada penemuan analog rapamycin (rapalog) termasuk Everolimus, Temsirolimus, dan Ridaforolimus, yang menunjukkan kelarutan air yang lebih baik, bioavailabilitas oral, dan fitur farmakokinetik lainnya [ [ 201 ] ]. Karena cara kerjanya yang tidak kompetitif terhadap ATP, rapalog telah menunjukkan profil yang relatif lebih aman [ [ 199 , 202 ] ], yang mengarah pada persetujuan klinisnya. Awalnya digunakan pada manusia sebagai obat imunosupresif untuk mencegah penolakan transplantasi, rapamycin dan analognya telah menunjukkan kemampuan untuk menginduksi autophagy dan mengerahkan fungsi neuroprotektif dalam beberapa model praklinis neurodegeneratif [ [ 134 , 203 – 209 ] ]. Mengingat bahwa rapalog menghambat mTORC1 secara tidak lengkap [ [ 210 ] ], penelitian selanjutnya difokuskan pada pengembangan inhibitor mTOR baru dengan efikasi yang ditingkatkan. Bertindak sebagai inhibitor kinase mTOR kompetitif ATP, inhibitor mTOR generasi kedua ini menargetkan domain kinase dari mTOR, sehingga menghambat kompleks mTORC1 dan mTORC2 [ [ 211 ] ]. Contoh penting dari inhibitor ini termasuk MLN0128, AZD2-14, OSI-027, WYE-354, Torkinib, dan beberapa di antaranya saat ini sedang dievaluasi dalam uji klinis untuk menilai profil toksisitas dan efikasi jangka panjang.
Terapi gen
Beberapa penelitian mengeksplorasi potensi terapi gen untuk peningkatan autofagi. Misalnya, transfer gen regulator autofagi utama TFEB ditemukan dapat memperbaiki patologi berbagai penyakit yang terkait dengan akumulasi agregat protein toksik intraseluler [ [ 212 ] ]. Transfer gen TFEB yang diarahkan ke hati pada model tikus penyakit hati yang terkait dengan akumulasi toksik mutan Z-alpha-1-antitrypsin (ATZ) mengakibatkan penurunan dramatis ATZ hati, apoptosis hati, dan fibrosis [ [ 176 ] ]. Dalam model tikus tauopati, ekspresi berlebihan TFEB secara efektif mengurangi patologi kekusutan neurofibrilar dan menyelamatkan defisit perilaku dan sinaptik [ [ 213 ] ]. TFEB juga mampu meningkatkan kinerja kognitif dan fungsi sinaptik dalam model penyakit Parkinson berdasarkan ekspresi berlebihan alfa-sinuklein manusia toksik di otak tengah tikus [ [ 214 ] ]. Yang penting, transfer gen TFEB menurunkan penyimpanan glikogen dan akumulasi berlebihan autofagosom dalam model tikus penyakit Pompe, suatu kelainan penyimpanan lisosomal [ [ 215 ] ]. Meskipun efek menguntungkan diamati dalam penelitian tersebut di atas, ekspresi berlebihan TFEB ditemukan menginduksi karsinoma sel ginjal [ [ 216 , 217 ] ], meningkatkan kekhawatiran tentang perkembangan klinisnya.
Contoh lain dari penelitian yang menunjukkan efek neuroprotektif yang menguntungkan setelah peningkatan autophagy melalui terapi gen didasarkan pada ekspresi berlebihan Becn1 dan Prkn , yang mendorong pembersihan protein salah lipat yang toksik pada model PD, AD, dan ataksia spinocerebellar tipe 3 [ [ 218 – 220 ] ]. Lebih jauh lagi, pengiriman vektor virus adeno-associated (AAV) yang mengekspresikan TRPML1 manusia , saluran Ca 2+ lisosomal yang mengatur autophagy melalui jalur TFEB dan VPS34 [ [ 8 , 221 ] ], ditemukan untuk mendorong pembersihan ATZ yang salah lipat dan mengurangi fibrosis hati pada model praklinis penyakit hati karena akumulasi ATZ [ [ 222 ] ].
Kesimpulan dan arah masa depan
Autofagi memainkan peran penting dalam menjaga homeostasis seluler dan mendukung banyak proses biologis, termasuk kontrol kualitas protein dan organel, diferensiasi dan perkembangan, imunitas, metabolisme, dan penuaan [ [ 223 – 228 ] ]. Oleh karena itu, disregulasi atau mutasi pada gen terkait autofagi telah terlibat dalam berbagai penyakit manusia. Dalam lanskap kanker yang kompleks, autofagi menunjukkan peran ganda: ia dapat bertindak baik sebagai kekuatan pro-tumorigenik dan sebagai penekan tumor. Menariknya, perubahan genetik yang secara langsung memengaruhi gen mesin autofagi jarang terjadi pada tumor manusia, menunjukkan bahwa hilangnya autofagi mungkin bukan kekuatan evolusi pendorong dalam fase inisiasi tumorigenesis. Lebih jauh lagi, pada pasien manusia yang terkena kanker, peningkatan kadar p62, protein yang sering dikaitkan dengan tumorigenesis, tidak muncul karena hilangnya autofagi (seperti yang terlihat pada beberapa model tikus) tetapi lebih sebagai akibat dari perolehan jumlah salinan kromosom 5q, tempat SQSTM1 (p62) berada. Oleh karena itu, hilangnya autofagi dan fungsi penekan tumornya memainkan peran yang terbatas, sedangkan aktivasi autofagi tampaknya lebih penting untuk tumorigenesis. Sel kanker secara strategis memanfaatkan autofagi untuk memastikan fleksibilitas metabolik, membentuk lingkungan mikro pro-inflamasi, menghindari pengawasan imun, dan memperoleh resistensi terhadap agen kemoterapi. Pengamatan ini menyoroti potensi terapeutik penargetan autofagi untuk pengobatan kanker, meskipun mengidentifikasi jenis tumor mana yang paling diuntungkan dari strategi tersebut memerlukan penyelidikan lebih lanjut. Dari perspektif klinis, sangat penting untuk menyeimbangkan kemanjuran dan keamanan modulasi autofagi, dengan mempertimbangkan peran pentingnya dalam homeostasis jaringan normal. Salah satu tantangan signifikan dalam mengembangkan modulator autofagi adalah kurangnya biomarker yang andal untuk memantau fluks autofagi dan keterlibatan target pada pasien, yang mempersulit pengoptimalan dosis dalam uji klinis fase awal. Penelitian di masa mendatang dapat memperdalam pemahaman kita tentang peran autofagi yang otonom pada berbagai jenis sel kanker, fungsinya dalam lingkungan mikro tumor, dan meningkatkan hasil terapeutik dengan inhibitor autofagi yang lebih kuat dan aman.
Pada kelainan konformasi protein, beberapa studi praklinis telah menunjukkan efek menguntungkan dari peningkatan autofagi untuk pembersihan agregat protein toksik. Hasil yang menjanjikan ini telah menyebabkan dimulainya uji klinis untuk menilai potensi peningkatan autofagi sebagai pendekatan terapeutik bagi pasien dengan kelainan tersebut. Namun, masih banyak pertanyaan terbuka yang belum terjawab. Misalnya, sebagian besar penyakit neurodegeneratif adalah bentuk sporadis yang patomekanisme yang mendasarinya tidak diketahui, kecuali untuk sebagian kecil kasus familial yang terkait dengan mutasi genetik yang diwariskan. Disfungsi mekanisme pembuangan protein, seperti autofagi dan sistem proteasom ubikuitin, berkontribusi secara signifikan terhadap patogenesis penyakit. Perlu dicatat, kemungkinan besar perubahan dalam jalur ini memiliki efek progresif, kumulatif, dan jangka panjang, yang mungkin menjadi jelas secara klinis di kemudian hari, karena banyak penyakit neurodegeneratif bermanifestasi selama akhir masa dewasa. Studi masa depan sangat menjanjikan untuk memajukan pemahaman kita tentang peran autofagi dalam penyakit. Ini termasuk mengidentifikasi regulator autofagi khusus penyakit yang baru dan mengembangkan strategi terapeutik baru untuk memanfaatkan autofagi untuk pembersihan agregat protein toksik.