
Abstrak
Peran relevan retrotransposisi LINE-1 (L1) dalam kanker telah berulang kali ditunjukkan dalam beberapa tahun terakhir. Namun, sifat retrotransposon yang berulang membuat identifikasi dan deteksinya tidak dapat diakses untuk praktik klinis. Selain itu, relevansi klinisnya bagi pasien kanker masih terbatas. Di sini, kami mengembangkan RetroTest, metode efisien baru untuk mengukur aktivasi L1 berdasarkan pengurutan yang ditargetkan dan jalur bioinformatika yang canggih, yang memungkinkan penerapannya dalam biopsi tumor. Pertama, kami melakukan pembandingan RetroTest untuk mengonfirmasi spesifisitas dan keandalannya yang tinggi. Kemudian, kami mengungkap aktivasi L1 dalam karsinoma sel skuamosa kepala dan leher (HNSCC) menurut kohort pasien yang luas yang mencakup semua stadium tumor. Estimasi retrotransposisi L1 mengungkapkan aktivasi yang sangat awal dalam perkembangan HNSCC, bertentangan dengan hubungan klasiknya dengan stadium lanjut. Selain itu, aktivasi L1 bersama dengan profil mutasi genomik pada jaringan normal yang berdekatan mendukung proses karsinogenesis lapangan, yaitu fenomena saat jaringan mengembangkan beberapa bercak sel dengan perubahan genetik dan/atau epigenetik, sehingga meningkatkan risiko perkembangan kanker di area tersebut. Secara keseluruhan, hasil kami menggarisbawahi aktivasi L1 dini pada HNSCC dan karakterisasi lapangan, yang menjadikan L1 sebagai biomarker diagnostik dini yang menjanjikan dan mendukung pentingnya estimasi retrotransposisi L1 dalam praktik klinis menuju diagnosis yang lebih efisien pada HNSCC.
Singkatan
FFPE
formalin-fiksasi parafin-tertanam
HNSCC
karsinoma sel skuamosa kepala dan leher
GARIS-1 (L1)
elemen nuklir yang tersebar panjang
Bahasa Indonesia: NAT
jaringan normal yang berdekatan
Sistem Operasi
kelangsungan hidup secara keseluruhan
PFS
kelangsungan hidup bebas perkembangan
PMBC
sel mononuklear darah tepi
TD2
transduksi L1 yatim piatu
TMB
beban mutasi tumor
Angkatan Udara
frekuensi alel varian
WGS
pengurutan genom secara keseluruhan
1 Pendahuluan
Sekitar setengah dari genom manusia tersusun atas elemen transposabel, urutan dengan kemampuan untuk berpindah dari satu lokasi ke lokasi lain, mengubah struktur normal genom di tempat-tempat di mana mereka terintegrasi [ [ 1 , 2 ] ]. Di antara mereka, retrotransposon elemen nuklir yang tersebar panjang (LINE-1, L1) mewakili 17% dari seluruh konten DNA dengan sekitar 500.000 salinan, sebagian besar terpotong atau tidak aktif [ [ 3 – 5 ] ]. Hanya sebagian kecil dari L1 yang tetap aktif dalam genom manusia, meskipun tetap ditekan secara transkripsi karena mekanisme epigenetik yang mencegah kerusakan yang akan disebabkan oleh mobilisasinya [ [ 6 ] ]. Ketika penekanan ini hilang, aktivasi L1 dapat menyebabkan berbagai penyakit, termasuk kanker [ [ 7 – 9 ] ]. Meskipun dampak aktivasi L1 yang ditunjukkan dalam genom kanker, sifatnya yang berulang dan penyebarannya di sepanjang genom menghalangi perkiraan aktivasi yang sebenarnya, mencegah penerapannya ke dalam praktik klinis.
Dalam kerangka Konsorsium Internasional Pan-Kanker (PCAWG), analisis kami sebelumnya menunjukkan bahwa insersi L1 somatik merupakan sumber restrukturisasi utama genom kanker, terutama penting untuk karsinoma sel skuamosa kepala dan leher (HNSCC), yang merupakan jenis tumor kedua dengan aktivasi L1 tertinggi [ [ 8 ] ]. Kanker kepala dan leher adalah kelompok kanker heterogen, yang lebih dari 90% didiagnosis sebagai HNSCC, yang timbul pada epitel berlapis rongga mulut, faring, dan laring [ [ 9 , 10 ] ]. Kurangnya gejala pada tahap awal, bersama dengan biomarker diagnostik yang tidak ada, menyebabkan sebagian besar diagnosis pada stadium lanjut, di mana tingkat kelangsungan hidup 5 tahun < 50% [ [ 10 ] ]. Dengan demikian, ada kebutuhan mendesak untuk menemukan biomarker molekuler yang dapat memfasilitasi diagnosis dini dan meningkatkan harapan hidup pasien.
Di sini, kami bertujuan untuk mengungkap dampak kemungkinan aktivasi L1 sepanjang perkembangan HNSCC dalam pengaturan klinis dengan mengembangkan metode baru yang efisien berdasarkan transduksi L1: RetroTest.
2 Bahan dan Metode
2.1 Pasien dan sampel tumor
Serangkaian 96 pasien HNSCC dari 3 kelompok berbeda ( Complexo Hospitalario Universitario de A Coruña (CHUAC), Biobanco Vasco , dan Fundación Pública Galega de Medicina Xenómica (FPGMX)) dianalisis. Semua pasien memahami dan menandatangani formulir persetujuan tertulis. Metodologi penelitian sesuai dengan standar yang ditetapkan oleh Deklarasi Helsinki. Metodologi penelitian disetujui oleh Komite Etik Penelitian Klinis setempat di Santiago–Lugo (CEIC 2018/567). Karakteristik ditentukan dalam Tabel 1 .
Tabel 1. Karakteristik dasar pasien HNSCC dan hasil klinikopatologis dalam seri tersebut.

2.2 Isolasi DNA
DNA genomik diekstraksi dari jaringan beku segar dan sampel yang difiksasi formalin dalam parafin (FFPE) menggunakan AllPrep DNA/RNA dan AllPrep FFPE DNA/RNA Mini Kits (Qiagen). Kuantifikasi dan integritas DNA dinilai menggunakan Qubit dsDNA BR Assay Kit dalam Qubit 4.0 (ThermoFisher) dan sistem TapeStation 4200 (Agilent).
2.3 Desain pustaka RetroTest dan pengurutan target
Ada tiga jenis retrotransposisi yang berbeda: solo-L1 (TD0), ketika L1 parsial atau lengkap diretrotransposisi; transduksi berpasangan (TD1), di mana L1 dan sekuens unik hilir diretrotransposisi; dan transduksi yatim piatu (TD2), di mana hanya sekuens unik hilir L1 aktif yang dimobilisasi tanpa L1 terkait [ [ 6 ] ]. Selama transkripsi L1, mesin transkripsi terkadang melewati sinyal poliadenilasi L1 hingga situs poliadenilasi 3′ hilir kedua, memobilisasi sekuens unik hilir elemen dalam proses yang disebut transduksi 3′ L1. Ini terjadi pada sekitar 10% mobilisasi L1 [ [ 6 ] ] dan dapat digunakan sebagai pengukuran tidak langsung dari aktivasi L1 nyata. RetroTest didasarkan pada sekuensing yang ditargetkan terhadap sekuens unik hilir dari 124 elemen kompeten panjang penuh L1 yang dijelaskan sebelumnya [ [ 6 ] ]. Gagasan utama di balik RetroTest adalah bahwa pasangan pembacaan yang tidak selaras, di mana salah satu pasangan dipetakan ke urutan hilir L1 3′ sementara yang lain dipetakan ke urutan target penyisipan, mendukung penyisipan. Selain itu, pembacaan yang terpotong, dipetakan ke urutan target tetapi berisi blatting ekstrem yang tidak selaras ke urutan hilir L1 3′, dideteksi untuk mengidentifikasi titik henti penyisipan. Dengan demikian, RetroTest mengidentifikasi penyisipan L1 melalui deteksi pembacaan yang tidak selaras dan pembacaan yang terpotong (Gbr. 1A ).
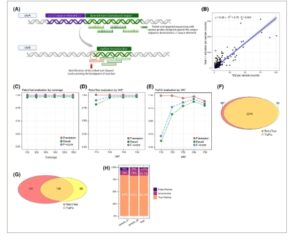
Gbr. 1
Buka di penampil gambar
Kekuatan Gambar
Desain dan pembandingan RetroTest. (A) Desain RetroTest. (B) Scatterplot dan korelasi antara aktivasi L1 dan transduksi orphan (TD2) dalam data International Consortium of PanCancer (PCAWG). Jumlah transduksi L1 3′ mengukur aktivasi L1 dalam 2.954 genom kanker dari 38 subtipe kanker histologis. (C) Kinerja RetroTest untuk cakupan sekuensing yang berbeda menggunakan genom hg19 yang dihasilkan secara artifisial dengan total 2.480 transduksi L1 yang didistribusikan secara acak pada 50% VAF. (D) Kinerja RetroTest sehubungan dengan VAF integrasi L1, menggunakan genom hg19 yang dihasilkan secara artifisial dengan total 2.480 transduksi L1 yang didistribusikan secara acak pada VAF yang berbeda. (E) Kinerja TraFiC berkenaan dengan VAF integrasi L1, menggunakan genom hg19 yang dihasilkan secara artifisial dengan total 2.480 transduksi L1 yang didistribusikan secara acak pada VAF yang berbeda. (F) Diagram Venn jumlah penyisipan L1 yang dideteksi oleh RetroTest dan TraFiC dalam genom artifisial dengan VAF 50% untuk penyisipan L1. (G) Diagram Venn jumlah penyisipan L1 yang dideteksi oleh RetroTest dan TrafiC dalam 19 sampel HNSCC yang menjalani WGS dan pengurutan target RetroTest. (H) Diagram batang peristiwa retrotransposisi positif benar, tidak meyakinkan, dan positif salah yang secara eksklusif dipanggil oleh RetroTest setelah pemeriksaan IGV dalam data WGS sampel HNSCC_21 dan sampel_28.
Pustaka sekuensing yang diperkaya dengan transduksi L1 3′ disiapkan mulai dari 100 ng DNA yang digunting menggunakan Covaris M220 Focused-Ultrasonicator (Covaris Inc.) dalam fragmen ~300 bp untuk FFPE dan ~500 bp untuk tumor beku. Setelah sonikasi, ukuran fragmen dan konsentrasi DNA dinilai dengan High Sensitivity DNA Assay (Agilent Technologies Inc.). Pustaka HNSCC yang diligasi adaptor disiapkan dengan SureSelect Target Enrichment System untuk Illumina Paired-End Multiplexed Sequencing menggunakan SureSelect XT2 Library Prep Kit (Agilent Technologies Inc.). Secara singkat, sampel diindeks, diperkuat, dan digabungkan sebelum hibridisasi dan penangkapan dengan umpan yang ditargetkan RNA. Kumpulan terindeks yang ditangkap diperkuat untuk mendapatkan pustaka akhir yang diperkaya. Kontrol kualitas dari persiapan pustaka dilakukan dengan menggunakan D1000 ScreenTape Assay dan High Sensitivity D1000 Assay (Agilent Technologies Inc.). Sampel multipleks diurutkan pada platform Illumina MiSeq dan NextSeq menggunakan pembacaan pair-end 150 bp dengan MiSeq Reagent Kit v2 dan NextSeq 500/550 Mid Output Kit v2.5 (300 Cycles) (Illumina Inc.). Cakupan rata-rata untuk sampel tumor adalah 101x, 726x untuk sampel normal yang berdekatan, dan 282x untuk sampel darah.
Pembacaan sekuensing dipetakan ke genom referensi hg19 oleh Burrows-Wheeler Aligner BWA-mem [ [ 11 ] ]. Samtools [ [ 12 ] ] digunakan untuk mengurutkan pembacaan yang selaras dan mengindeks file bam yang diperoleh, menerapkan Bammarkduplicates2 dari alat Picard [ [ 13 ] ] untuk menandai pembacaan duplikat.
2.4 Alur kerja RetroTest
Input RetroTest diproses dengan file BAM dengan pembacaan 150 bp, yang berasal dari pengurutan berpasangan Illumina. Identifikasi klaster pendukung penyisipan dilakukan sebagai berikut:
Peristiwa pembacaan yang tidak selaras dan terpotong dicari dalam berkas BAM. Langkah ini dapat dilakukan dalam a. mode sampel tunggal atau b. mode pencocokan tumor-normal jika kontrol germline disediakan. Kemudian, peristiwa pembacaan terpotong kiri dan kanan disejajarkan kembali untuk mencari penyelarasan tambahan.
Pembacaan diskordan dan kliping disusun ke dalam wadah genomik lalu dikelompokkan ke dalam kluster. Kemudian pasangan pembacaan diskordan dikelompokkan berdasarkan posisi pasangan sementara pembacaan kliping dikelompokkan berdasarkan posisi penyelarasan suplementer. Wadah genomik untuk mencari insersi (yang sesuai dengan area transduksi) didasarkan pada koordinat daerah transduksi hilir L1.
Untuk penyaringan klaster, semua klaster tanpa jumlah minimum pembacaan pendukung, yang terletak di wilayah yang tidak spesifik, atau yang terdiri dari pembacaan duplikat dibuang. Klaster juga dapat disaring berdasarkan koordinat genomik yang diberikan dan kualitas pemetaan penyelarasan rata-rata. Selain itu, untuk klaster yang tidak selaras, klaster dengan pasangan yang tidak berada di atas referensi target, dan klaster yang pasangannya berada di atas wilayah hilir elemen sumber mana pun dibuang. Untuk klaster yang terpotong, klaster dengan penyelarasan tambahan di luar referensi target dan klaster yang penyelarasan tambahannya memetakan wilayah hilir elemen sumber mana pun juga disaring.
Klaster-klaster yang tidak selaras dan terpotong yang telah disaring dikelompokkan ke dalam metaklaster, yang koordinat tepatnya ditentukan oleh titik henti metaklaster.
Akhirnya, setiap jenis transduksi metakluster ditentukan menggunakan pembacaan diskordan di sekitar titik penyisipan, berdasarkan posisi pemetaan pasangan jangkar.
Pembacaan pendukung dapat diselaraskan secara eksklusif dengan untaian positif dan dilaporkan sebagai gugus PLUS, secara eksklusif dengan untaian negatif dan dilaporkan sebagai MINUS, atau dengan kedua untaian dan dilaporkan sebagai gugus TIMBAL BALIK.
Saluran tersebut dapat ditemukan di situs berikut: https://gitlab.com/mobilegenomesgroup/RETROTEST .
2.5 Pembandingan RetroTest
Kami membandingkan kinerja RetroTest dengan Transposon Finder in Cancer (TraFiC), yang digunakan sebelumnya di PCAWG [ [ 6 ] ], dengan menghasilkan simulasi kejadian dan kondisi.
Untuk mensimulasikan transduksi yatim piatu, 5Kb urutan genom referensi hilir (hg19) diambil untuk masing-masing dari 124 elemen sumber yang disertakan dalam basis data MEIGA-MEIsimulator [ [ 14 ] ], setelah mempertimbangkan orientasi L1. Untuk setiap elemen sumber, 20 urutan transduksi dihasilkan oleh pemangkasan 3′ acak, yang mewakili akhir transkripsi alternatif. Untuk mensimulasikan peristiwa penyisipan, urutan genom referensi dibagi menjadi tempat sampah 10 kb, dan titik penyisipan dipilih secara acak di antara tempat sampah yang terdapat dalam kromosom nuklir, dengan satu-satunya kondisi menghindari daerah GAP. Urutan DNA dari genom referensi dan transduksi yatim piatu digabungkan secara berurutan menggunakan perintah python khusus, sambil menambahkan fitur karakteristik MEI, termasuk duplikasi situs target, ekor poliA, dan pemotongan 5′.
Kami menghasilkan pembacaan ujung berpasangan in silico dari versi genom yang dimodifikasi ini dengan ART v2.5.8 [ [ 15 ] ] melalui beberapa perintah dari MEIGA-MEIsimulator, alat bioinformatika internal (panjang 150 bp, ukuran sisipan 350 bp ± 10%), yang kemudian diselaraskan dengan BWA- mem v0.7.17 [ [ 11 , 16 ] ] terhadap genom referensi hg19 dan diproses lebih lanjut dengan samtools v1.3.1 [ [ 12 , 16 ] ].
Kami memilih pasangan bacaan dengan setidaknya satu pasangan memetakan pada kumpulan wilayah target tertentu dengan Picard FilterSamReads v2.18.14 [ [ 13 ] ]. Kami menggunakan berkas bam yang dihasilkan untuk menilai sensitivitas dan spesifisitas dalam kondisi yang berbeda. Untuk menguji kinerja metode kami dengan peristiwa subklonal, kami mensimulasikan transduksi pada VAF yang berbeda (10%, 20%, 40% dan 50%). Kami juga mempelajari bagaimana cakupan memengaruhi daya deteksi algoritme kami. Menggunakan Picard DownSampleSam v2.18.14 [ [ 13 ] ], kami mengambil subsampel bacaan dari simulasi 150x pada 50% VAF di bawah kedalaman sekuensing yang berbeda (15x, 30x, 60x, 90x, 120x, dan 150x).
Presisi dihitung dengan membagi jumlah panggilan positif benar dengan jumlah total panggilan. Penarikan kembali dihitung dengan membagi jumlah panggilan positif benar dengan jumlah total kejadian yang disimulasikan. Positif benar, positif palsu, dan negatif palsu diidentifikasi dengan memotong koordinat kejadian yang disimulasikan dengan koordinat panggilan menggunakan BEDTools [ [ 17 ] ].
Selain itu, Trafic dan RetroTest dibandingkan dalam subset sampel HNSCC yang menjalani WGS (lihat Metode 2.6) dan metode target RetroTest. Diagram Venn dengan insersi yang dideteksi oleh setiap metode diplot menggunakan paket vennDiagram R. Akhirnya, untuk dua sampel HNSCC, semua panggilan L1 TD2 khusus RetroTest diperiksa secara visual dengan Integrative Genomics Viewer (IGV) dalam data WGS. Untuk mengklasifikasikan insersi kandidat sebagai retrotransposisi somatik sejati, kriteria berikut harus dipenuhi: (i) setidaknya tiga bacaan pendukung dalam data WGS Illumina tumor; (ii) tidak ada bacaan pendukung dalam data WGS Illumina normal yang cocok; dan (iii) keberadaan setidaknya satu ciri retrotransposisi, baik duplikasi situs target (TSD) atau ekor poli(A/T), dengan titik henti yang ditentukan secara tepat.
2.6 Analisis genom keseluruhan, profil mutasi, dan analisis pengayaan
Untuk WGS, Truseq Nano DNA Libraries (350 bp) dibangun dan diurutkan dalam platform NovaSeq6000 Illumina (150 bp paired-end) dalam layanan eksternal (Macrogen). Pembacaan sekuensing dari jaringan tumor dan jaringan normal yang berdekatan (NAT) dipetakan ke genom referensi hg19 oleh Burrows-Wheeler Aligner BWA- mem [ [ 11 , 16 ] ] v0.7.17. samtools [ [ 12 , 18 ] ] v1.9 digunakan untuk mengurutkan pembacaan yang selaras dan mengindeks file bam yang diperoleh, menerapkan Picard Bammarkduplicates2 [ [ 13 ] ] untuk menandai pembacaan duplikat. Setelah itu, Mutect2 [ [ 19 ] ], dari Genome Analysis Tool Kit (GATK) [ [ 20 ] ] v4.1.1.0, digunakan untuk melakukan pemanggilan SNV dan INDEL. Varian difilter dengan FilterMutectCalls (GATK) (mempertimbangkan estimasi kontaminasi normal ASCAT [ [ 21 ] ]), mengikuti ambang batas standar, dan diberi anotasi menggunakan Ensembl Variant Effect Predictor (VEP) [ [ 22 ] ] v100.2. Kami memilih varian yang mungkin patogenik tersebut, mengikuti anotasi SIFT [ [ 23 , 24 ] ], PolyPhen [ [ 25 ] ], dan VEP Impact. Kemudian, kami memastikan varian kami bersifat somatik dengan memfilter varian dengan frekuensi alel yang sama dengan atau lebih tinggi dari 0,01 pada populasi 1KGP, ESP, atau genomAD, dengan menilai bahwa varian tersebut tidak umum dalam populasi.
Untuk menganalisis profil mutasi NAT, kami juga mengurutkan darah untuk 4 pasien. Kami kembali menggunakan mutect2 v4.1.7.0 untuk melakukan SNV gabungan dan INDEL yang memanggil tumor dan NAT, mengikuti praktik terbaik [ [ 26 , 27 ] ]. Dalam kasus ini, GATK FilterMutectCalls sekarang mempertimbangkan estimasi kontaminasi lintas sampel yang dilakukan oleh GATK CalculateContamination. Kami menganggap sebagai kesamaan antara NAT dan tumor varian yang menyajikan lebih dari satu bacaan pendukung di kedua sampel, sementara yang eksklusif untuk NAT menyajikan lebih dari satu bacaan pendukung dalam sampel ini dan tidak ada di tumor.
Analisis pengayaan dilakukan dengan paket R enrichr [ [ 28 ] ] menggunakan basis data referensi berikut: koleksi Human MSigDB [ [ 29 , 30 ] ] (MSigDB_Hallmark_2020, MSigDB_Oncogenic_Signatures, dan MSigDB_Computational) BioPlanet_2019 [ [ 31 ] ], KEGG_2019_Human [ [ 32 , 33 ] ], WikiPathways_2019_Human [ [ 34 , 35 ] ], GO_Molecular_Function_2018, dan GO_Biological_Process_2018 [ [ 36 , 37 ] ]. Kami menggunakan database motif pengikatan faktor transkripsi berikut: TRANSFAC_and_JASPAR_PWMs [ [ 38 ] ] dan ChEA_2016 [ [ 28 , 39 ] ].
2.7 Analisis statistik
Hubungan antara transduksi L1 (dikoreksi oleh cakupan) dan fitur klinis pasien dinilai dengan regresi linier berganda. Uji Wilcoxon atau Fisher, tergantung pada ukuran sampel, diterapkan. Analisis kelangsungan hidup menyeluruh (OS), kelangsungan hidup bebas progresi (PFS), dan probabilitas kelangsungan hidup dilakukan dengan paket survminer dan survival R, menggunakan uji log-rank. Hubungan antara kelangsungan hidup dan variabel klinis dievaluasi dengan regresi Cox.
3 Hasil
3.1 Pembandingan RetroTest
RetroTest dirancang untuk menangkap sekuens unik yang dimobilisasi dan ditransduksi hilir dari transduksi L1 yatim piatu (TD2), digunakan sebagai kode batang (Gbr. 1A ). Kode batang ini dapat mengidentifikasi dengan tegas penyisipan yang disebabkan oleh 124 elemen sumber L1 yang aktif dalam kanker (Tabel S1 ) [ [ 6 , 8 ] ]. Dengan demikian, kami memfokuskan desain probe RetroTest pada 5.000 nukleotida pertama yang berdekatan dengan daerah 3′ L1 untuk setiap elemen sumber L1, sebagai daerah yang paling sering ditransduksi [ [ 6 ] ]. Karena TD2 digunakan sebagai ukuran tidak langsung dari total aktivasi L1, kami pertama-tama membandingkan frekuensi kedua kejadian, menemukan korelasi linier yang kuat dengan rasio 1:6 ( y = 6,33x , R2 = 0,78, P < 0,001, Gbr. 1B ). Berikutnya, kami mengoptimalkan protokol laboratorium untuk FFPE dan jaringan beku segar dan mengembangkan jalur bioinformatika terkait. Kami mengevaluasi kinerja dan akurasi RetroTest dengan membuat genom kanker buatan, tempat kami mendistribusikan secara acak total 2.480 transduksi L1. Menggunakan simulasi untuk kedalaman sekuensing yang berbeda, RetroTest memperoleh presisi sekitar 0,99 dalam semua kasus dan penarikan kembali sekitar 0,96 (Gbr. 1C ). Kami juga mengevaluasi kinerja tergantung pada frekuensi alel varian (VAF): RetroTest memperoleh presisi sekitar 1, menurun untuk VAF yang lebih rendah, dan penarikan kembali berkisar dari 0,81 hingga 0,96, meningkat seiring dengan peningkatan VAF (Gbr. 1D ).
Kemudian, kami membandingkan RetroTest dan TraFiC klasik, yang digunakan dalam PCAWG [ [ 6 ] ]. Karena TraFiC dirancang untuk bekerja hanya dengan data WGS 30x standar, kami membandingkan analisisnya, dengan memvariasikan VAF secara eksklusif. Presisi TraFiC berkisar dari 0,99 hingga 0,88 dan recall berkisar dari 0,11 hingga 0,87 saat VAF meningkat (meskipun recall maksimum diperoleh untuk VAF sebesar 40%, yaitu 0,87) (Gbr. 1E ) (Tabel S2 ). Kami membandingkan kinerja kedua metode dengan memotong kedua panggilan dalam genom tumor tiruan. Menggunakan VAF sebesar 50%, sebagian besar varian dipanggil oleh kedua metode, secara konkret 2.216, sementara RetroTest secara eksklusif memanggil 167, dan TraFiC 33 kejadian privat, sebagian besar menghasilkan positif palsu menurut IGV (Gbr. 1F ).
Untuk lebih memvalidasi alat kami, kami membandingkan kejadian TD2 yang diidentifikasi oleh RetroTest dan TraFiC dalam subset 19 sampel HNSCC yang menjalani WGS dan pengurutan target, menemukan bahwa 38,3% (136/355) kejadian terdeteksi oleh kedua metode, sementara 42,5% (151/355) diidentifikasi secara eksklusif oleh RetroTest, dan 19,2% (68/355) dideteksi secara eksklusif oleh TraFiC (Gbr. 1G , Tabel S3 ).
Pemeriksaan IGV atas kejadian TD2 yang secara eksklusif dipanggil oleh RetroTest pada data WGS dari sampel tumor HNSCC sample_21 (32 kejadian eksklusif RetroTest) dan sample_28 (52 kejadian eksklusif RetroTest) (Tabel S3 ) mengonfirmasi bahwa 83,3% dari retrotransposisi privat RetroTest adalah kejadian positif sejati (70 dari 84), karena kejadian tersebut menunjukkan sedikitnya tiga pembacaan pendukung dalam tumor, tidak ada pembacaan pendukung dalam normal yang cocok, dan menunjukkan ciri khas retrotransposisi (Gbr. S1 ). 13,1% sesuai dengan hasil yang tidak meyakinkan (11 dari 84), di mana tidak ada pembacaan yang dapat mengonfirmasi kejadian positif sejati, tetapi tidak ada bukti di wilayah tersebut yang menunjukkan positif palsu (menunjukkan bahwa ini kemungkinan kejadian L1 subklonal yang hanya ada di pustaka RetroTest). Akhirnya, hanya 3,6% yang positif palsu (3 dari 84 kejadian) (Gbr. 1H , Data S1 ).
3.2 Aktivasi L1 pada HNSCC
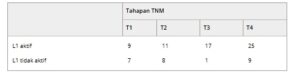
Setelah mengonfirmasi keakuratan dan presisi RetroTest, kami memutuskan untuk menerapkannya pada 96 kohort HNSCC dari stadium T1 hingga T4 (Tabel 1 ). Kami mendeteksi aktivasi L1 pada 71,8% pasien (Gbr. 2A , Tabel S4 ), yang mana 49,3% menunjukkan aktivitas tinggi (di luar median) (Tabel 2 ). Ketika aktivasi dipelajari menurut stadium tumor, penyakit lanjut (T3–T4) menunjukkan aktivasi L1 yang secara statistik lebih tinggi dibandingkan dengan stadium awal (T1–T2) ( P = 0,0051) (Gbr. 2B ). Secara khusus, aktivasi L1 terdeteksi pada 56,2% tumor T1, 57,9% T2, 94,4% T3, dan 73,5% tumor T4 (Tabel 2 ). Deteksi aktivasi L1 di semua stadium tumor, bahkan pada stadium pertama, menunjukkan aktivasi L1 dini.
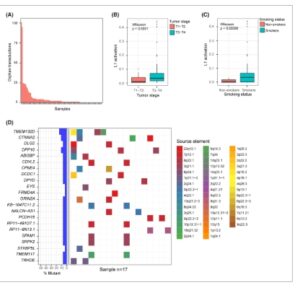
Gambar 2
Buka di penampil gambar
Kekuatan Gambar
Aktivasi L1 diukur dengan RetroTest dalam kohort HNSCC ( n = 96). (A) Kuantifikasi aktivasi L1 pada tumor HNSCC sebagai jumlah transduksi yatim piatu yang dideteksi oleh RetroTest. (B) Boxplot aktivasi L1 sehubungan dengan stadium TNM awal (T1–T2) dan lanjut (T3–T4). Nilai- P aktivasi diferensial diperoleh dengan uji Wilcoxon. Kumis boxplot meluas ke nilai terkecil dan terbesar dalam 1,5 kali rentang interkuartil kuartil bawah dan atas (Q1–Q3). Nilai outlier di luar rentang ini diplot sebagai titik individual. Untuk mengoreksi bias terkait cakupan, aktivasi L1 dihitung sebagai jumlah TD2 dibagi dengan cakupan mediannya. Sampel T1–T2 ( n = 35), sampel T3–T4 ( n = 52). (C) Boxplot aktivasi L1 sehubungan dengan status merokok. Nilai- P aktivasi diferensial diperoleh dengan uji Wilcoxon. Kumis boxplot meluas ke nilai terkecil dan terbesar dalam 1,5 kali rentang interkuartil kuartil bawah dan atas (Q1–Q3). Nilai outlier di luar rentang ini diplot sebagai titik individual. Untuk mengoreksi bias terkait cakupan, aktivasi L1 dihitung sebagai jumlah TD2 dibagi dengan cakupan mediannya. Perokok ( n = 64), bukan perokok ( n = 22). (D) Oncoplot yang menunjukkan gen yang dipengaruhi oleh insersi L1 dan elemen sumber aslinya. Sebanyak 17 pasien menunjukkan lebih dari 2 gen yang dipengaruhi oleh insersi L1 dari 46 elemen sumber yang berbeda.
Tabel 2. Jumlah pasien HNSCC yang menunjukkan aktivasi L1 sepanjang stadium TNM.
Dengan mempertimbangkan karakteristik klinis yang berbeda, kami tidak menemukan hubungan yang signifikan secara statistik antara aktivasi L1 dan konsumsi alkohol ( P = 0,14) atau jenis kelamin ( P = 0,055). Pasien perokok menunjukkan aktivitas L1 yang secara statistik signifikan lebih tinggi daripada bukan perokok ( P = 0,00098) (Gbr. 2C ). Tidak ada hubungan antara aktivasi L1 dan probabilitas kelangsungan hidup yang terdeteksi ( P = 0,86 aktif vs. tidak aktif, P = 0,28 tingkat tinggi vs. rendah) (Gbr. S2 ). Ketika hanya mempertimbangkan pasien T1, mereka dengan aktivasi L1 menunjukkan tren potensial dari probabilitas kelangsungan hidup yang berkurang meskipun tidak mencapai signifikansi statistik ( P = 0,77).
Akhirnya, kami mengidentifikasi transduksi L1 di dalam gen; secara konkret, kami menemukan 22 gen yang mengandung transduksi pada setidaknya dua pasien dari 17 pasien. Karena RetroTest dapat mengidentifikasi elemen sumber L1, kami mengungkap beberapa elemen sumber yang sangat aktif dalam HNSCC, terutama 22q12.1 (Gbr. 2D ).
3.3 Profil mutasi HNSCC dan aktivasi L1
Kami memperoleh data WGS dari 19 sampel tumor yang diukur juga oleh RetroTest. Untuk mendeteksi hanya variasi somatik, sampel normal berpasangan yang sesuai disertakan sebagai kontrol germline. Kami mendeteksi median 40 varian nukleotida tunggal (SNV) dan INDEL, mengidentifikasi total 1012 varian somatik, yang memengaruhi 918 gen (Tabel S5 ). Kami tidak mendeteksi korelasi antara aktivasi L1 dan beban mutasi tumor umum (TMB) (Gbr. 3A ). Hasil kami menunjukkan bahwa gen yang paling sering bermutasi adalah TP53 (36,8%), diikuti oleh NOTCH1 (26,3%), MT-ND5 (26,3%), FAT1 , dan GRIN2A (21,1%). Menariknya, kami menemukan bahwa sebagian besar pasien dengan mutasi TP53 juga menunjukkan aktivitas L1 yang tinggi (71,4%). Faktanya, ketika kami membandingkan aktivasi L1 dengan mutasi TP53 , kami menemukan kecenderungan asosiasi ( P = 0,11) (Gbr. 3B ). Analisis pengayaan dengan gen yang bermutasi menunjukkan proses kunci yang terlibat untuk perkembangan kanker seperti pensinyalan Notch , pensinyalan TGFß , dan lagi regulasi aktivitas p53 (Gbr. 3C ) (Tabel S6 ) dan perubahan faktor transkripsi yang terkait dengan mekanisme epigenetik termasuk Polycomb ( EZH2 , SUZ12 ) (Gbr. 3D ) (Tabel S7 ).

Gambar 3
Buka di penampil gambar
Kekuatan Gambar
Karakterisasi profil mutasi pasien HNSCC dengan WGS ( n = 19). (A) Oncoplot yang menunjukkan gen yang mengandung mutasi somatik pada pasien HNSCC. Jenis mutasi pada setiap gen dan aktivasi L1 (jumlah insersi L1 pada setiap pasien) ditunjukkan. (B) Boxplot aktivasi L1 sehubungan dengan mutasi TP53 atau status tipe liar. Kumis boxplot meluas ke nilai terkecil dan terbesar dalam 1,5 kali rentang interkuartil kuartil bawah dan atas (Q1–Q3). Nilai outlier di luar rentang ini diplot sebagai titik individual. Untuk mengoreksi bias terkait cakupan, aktivasi L1 dihitung sebagai jumlah TD2 dibagi dengan cakupan mediannya. Nilai- P aktivasi diferensial diturunkan dari uji Wilcoxon. TP53 bermutasi ( n = 7), WT ( n = 12). (C) Barplot analisis pengayaan Pathway berdasarkan 918 gen yang bermutasi secara somatik. Nilai P pengayaan dihitung dengan uji eksak Fisher. (D) Grafik batang analisis pengayaan pengikatan faktor transkripsi berdasarkan 918 gen yang bermutasi secara somatik. Nilai P pengayaan dihitung dengan uji eksak Fisher.
3.4 Profil jaringan normal yang berdekatan dengan tumor HNSCC (NAT)
Kami memutuskan untuk mengevaluasi kemungkinan proses karsinogenesis lapangan, di mana populasi sel normal digantikan oleh sel-sel yang disiapkan untuk kanker, tanpa perubahan anatomi atau morfologi, tetapi sudah premaligna pada tingkat molekuler. Kami menganalisis sampel normal yang tersedia yang diperoleh dari NAT, dengan sel mononuklear darah tepi (PBMC) digunakan sebagai kontrol germline (Gbr. 4A ). Kami mendeteksi total 25 varian somatik berdampak tinggi dan/atau mungkin patogen yang memengaruhi NAT (Tabel S8 ). Sebagian besar mutasi spesifik ini ( n = 20; 80%) eksklusif untuk NAT, termasuk yang memengaruhi gen kunci seperti NOTCH1 (gen yang paling bermutasi), FAT1 , atau PPARD ; sementara 20% dibagi antara NAT dan jaringan tumor, yang memengaruhi gen seperti CDKN2A .
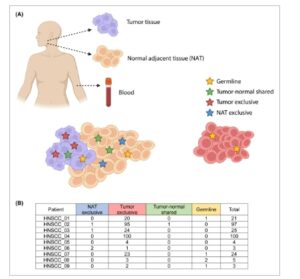
Gambar 4
Buka di penampil gambar
Kekuatan Gambar
Evaluasi aktivasi L1 pada jaringan normal yang berdekatan. (A) Representasi skematis dari evaluasi proses karsinogenesis lapangan. (B) Jumlah elemen aktif L1 pada jaringan normal yang berdekatan dari pasien HNSCC ( n = 9), dibandingkan dengan aktivasi L1 pada jaringan tumor berpasangan dan PBMC sebagai kontrol garis keturunan dari pasien yang sama.
Untuk menjelaskan apakah L1 lebih lanjut mendukung karsinogenesis lapangan, kami mengevaluasi 9 NAT dengan RetroTest dan membandingkan elemen L1 yang aktif dalam tumor, dalam NAT, dan dalam germline berpasangan yang sesuai. Kami dapat mengonfirmasi bahwa sebagian besar aktivasi L1 hanya ada di tumor; 5 insersi menghasilkan germline, dan 1 elemen dibagi oleh tumor dan NAT. Anehnya, 4 insersi muncul secara eksklusif di NAT (Gbr. 4B ). Dengan demikian, kami dapat mengonfirmasi karsinogenesis lapangan dan menunjukkan bahwa L1 sudah aktif di NAT, mendukung sekali lagi aktivasi awal di HNSCC.
4 Diskusi
Sejak dampak tinggi L1 yang baru-baru ini ditunjukkan dalam genom kanker [ [ 6 , 8 ] ], L1 telah dievaluasi sebagai biomarker kanker dalam berbagai penelitian yang menilai aktivitasnya dengan berbagai pendekatan berdasarkan tingkat ekspresi [ [ 40 – 43 ] ]. Namun, sebagian besar sekuens L1 terpotong dan tidak berfungsi, sehingga hasilnya dapat menyajikan bias yang penting, sementara penerjemahannya ke dalam rutinitas klinis menantang karena ketidakstabilan RNA/protein. Teknologi lain yang diusulkan didasarkan pada sekuensing penangkapan L1 DNA sepanjang penuh [ [ 44 ] ], tetapi menyajikan bias yang sama yang terkait dengan aktivasi L1 nyata dan persyaratan DNA input yang penting, tidak terjangkau untuk praktik klinis, selain tidak mengidentifikasi elemen L1 mana yang aktif dan seberapa banyak masing-masing berkontribusi pada aktivasi. Pendekatan terbaru didasarkan pada identifikasi insersi L1 sebagai ukuran aktivasi L1 nyata (TraFiC [ [ 6 ] ], xTea [ [ 45 ] ], MELT [ [ 46 ] ] dan Mobster [ [ 47 ] ]) menggunakan Whole Genome atau Whole Exome Sequencing, yang tidak terjangkau bagi sebagian besar rumah sakit. Selain itu, pendekatan ini didasarkan pada pembacaan pendek, yang menghambat deteksi insersi di daerah penataan ulang yang sangat berulang atau kompleks. Baru-baru ini, pengurutan pembacaan panjang telah muncul (xTea [ [ 45 ] ], PALMER [ [ 48 ] ]), tetapi sekali lagi persyaratan input DNA mereka tetap tidak terjangkau bagi sebagian besar biopsi klinis. Dengan demikian, menggabungkan aktivasi L1 ke dalam praktik klinis memerlukan metode standar baru.
Kami memperkenalkan RetroTest: metode baru untuk mendeteksi aktivasi L1 nyata dalam sampel klinis dengan persyaratan input DNA rendah, baik dari biopsi segar/beku maupun biopsi FFPE. Kebaruan dan kekuatannya terletak pada kemampuannya mengidentifikasi secara pasti insersi yang disebabkan oleh 124 elemen L1 sumber L1 yang aktif dalam kanker dengan cara yang lebih hemat biaya daripada pendekatan yang diusulkan sebelumnya. Metode kami tidak hanya menawarkan estimasi L1 global tetapi juga mengidentifikasi elemen sumber L1 yang aktif. Pembandingan kami mendukung presisi dan perolehan kembali yang tinggi untuk RetroTest, bahkan mendeteksi insersi subklonal. Kesamaan yang tinggi dengan TraFic, baik dalam simulasi maupun data dunia nyata, mendukung potensi tinggi untuk metodologi baru ini.
Retrotransposisi somatik L1 adalah jenis varian struktural kedua yang paling sering terjadi pada genom HNSCC [ [ 8 ] ]. Sesuai dengan itu, kami mendeteksi bahwa sebagian besar pasien HNSCC (75%) menunjukkan aktivasi L1, hampir setengahnya dengan kadar yang tinggi. Aktivasi ini lebih tinggi pada stadium lanjut, seperti yang dijelaskan pada esofagus Barret, di mana aktivitas L1 yang lebih rendah muncul pada stadium awal, meningkat seiring dengan perkembangan kanker [ [ 49 ] ]. Kami menemukan aktivasi yang sangat awal pada stadium awal HNSCC, dengan aktivasi L1 pada 62–63% tumor T1. Data ini menunjukkan aktivasi L1 sebagai peristiwa awal dalam konfigurasi genom HNSCC dan, dengan demikian, dalam perkembangan penyakit. Faktanya, pasien T1 dengan aktivasi L1 cenderung menunjukkan prognosis yang lebih buruk.
Kami menemukan hubungan antara aktivitas L1 yang tinggi dan kebiasaan merokok. Penelitian sebelumnya telah melaporkan tingkat hipometilasi L1 yang lebih tinggi pada perokok [ [ 50 ] ]. Karena 75% HNSCC dikaitkan dengan tembakau [ [ 51 ] ], mekanisme ini dapat bertanggung jawab atas tingginya tingkat aktivitas L1, karena ini merupakan mekanisme yang paling terbukti mencegah reaktivasi L1 [ [ 6 , 52 , 53 ] ]. Faktanya, hipometilasi L1 telah dikaitkan dengan prognosis yang lebih buruk pada HNSCC [ [ 54 , 55 ] ] dan ditemukan sebagai kejadian awal pada kanker kolorektal, lambung, dan mulut [ [ 56 – 58 ] ].
Elemen sumber L1 yang paling aktif dalam kelompok kami berada di 22q12.1, bertepatan dengan hasil sebelumnya [ [ 10 , 59 , 60 ] ]. L1 ini telah diidentifikasi sebagai mRNA L1 utuh yang paling banyak diekspresikan pada payudara, ovarium, dan CRC [ [ 61 , 62 ] ], dan elemen L1 yang menjelaskan sebagian besar transduksi pada CRC [ [ 52 ] ], yang menunjukkan aktivitas terpanas yang sama pada HNSCC.
Kami tidak menemukan korelasi antara aktivitas L1 dan TMB, tetapi ada hubungan antara mutasi TP53 dan aktivasi L1, sejalan dengan penelitian sebelumnya yang menunjukkan bahwa TP53 dapat menekan mobilisasi L1 [ [ 8 , 61 , 63 , 64 ] ]. Kami juga mengidentifikasi regulasi epigenetik, terutama yang terkait dengan kompleks represif Polycomb. Menariknya, Mangoni et al. telah menguraikan bahwa RNA L1 dapat bertindak sebagai RNA non-coding panjang dan berinteraksi langsung dengan Polycomb selama perkembangan dan evolusi otak [ [ 65 ] ]. Kami sebelumnya melaporkan bahwa Polycomb dapat mengatur lncRNA HOTAIR pada kanker kandung kemih [ [ 66 ] ] dan Ishak et al. menunjukkan pembungkaman urutan pengulangan genom yang bergantung pada EZH2, termasuk elemen L1 [ [ 67 ] ]. Oleh karena itu, analisis tambahan diperlukan untuk lebih lanjut membahas apakah jaringan epigenetik ini berperan dalam reorganisasi genom kanker.
Kami mengevaluasi keberadaan kankerisasi medan, menemukan mutasi somatik eksklusif di NAT dan sebagian kecil yang dibagi dengan tumor. Beberapa penelitian telah menggambarkan tambal sulam klon yang berbeda dalam jaringan normal, bahkan mengandung mutasi penggerak [ [ 68 , 69 ] ]. Kami menemukan NOTCH1 dan FAT1 sebagai gen yang paling bermutasi, seperti hasil Martincorena pada kulit dan esofagus normal [ [ 68 , 70 ] ], yang menunjukkan keberadaan medan invasi prakanker atau kanker. Akhirnya, beberapa mobilisasi L1 secara eksklusif ditemukan di NAT, dan hanya satu yang tampak dibagi dengan tumor, sekali lagi mendukung medan kankerisasi. Retrotransposisi L1 somatik juga dideskripsikan pada urothelium dan epitel kolorektal normal, meskipun dengan tingkat yang jauh lebih rendah dibandingkan pada kanker [ [ 71 – 73 ] ], dan pada sampel pra-tumor termasuk esofagus Barrett dan adenoma kolorektal [ [ 49 , 59 , 74 , 75 ] ].
5 Kesimpulan
Kesimpulannya, aktivasi L1 muncul sebagai peristiwa awal yang diperkuat dalam sejarah alami HNSCC, bahkan pada tahap pra-tumor, dengan potensi tinggi sebagai biomarker kanker lapangan dan diagnosis dini HNSCC.
Ucapan Terima Kasih
Penulis mengucapkan terima kasih kepada semua pasien yang terdaftar dan keluarga mereka. Proyek penelitian ini dimungkinkan melalui akses yang diberikan oleh Galician Supercomputing Center (CESGA) ke infrastruktur superkomputernya. Superkomputer FinisTerrae III dan sistem penyimpanan data permanennya telah didanai oleh Kementerian Sains dan Inovasi Spanyol, Pemerintah Galisia, dan Dana Pembangunan Regional Eropa (ERDF). Pekerjaan ini telah didanai oleh Instituto de Salud Carlos III (ISCIII) melalui proyek PI19/01113 dan didanai bersama oleh Uni Eropa, dan oleh Spanish Association Against Cancer Scientific Foundation (IDEAS19122MART). AO didukung oleh beasiswa predoktoral dari Galician Innovation Agency, Xunta de Galicia (ED481A-2020/214). MM-F dan JB sebelumnya didukung oleh Spanish Association Against Cancer Scientific Foundation (masing-masing INVES207MART dan PRDCR19007BREA_001). MM-F saat ini didukung oleh program Miguel Servet (CP20/00188) dari Instituto de Salud Carlos III (ISCIII) dan Dana Sosial Eropa (“Berinvestasi untuk masa depan Anda”). MG-G didukung oleh beasiswa pascadoktoral dari Badan Inovasi Galisia, Xunta de Galicia (IN606B-2024/014).